最近收到读者求助,说他感兴趣的表达量芯片数据集用到的的芯片是:[HT_HG-U133_Plus_PM] Affymetrix HT HG-U133+ PM Array Plate ,看起来跟我们授课的 hg133plus2比较类似。
但是很明显,看主页信息,一点都不简单 https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GPL13158
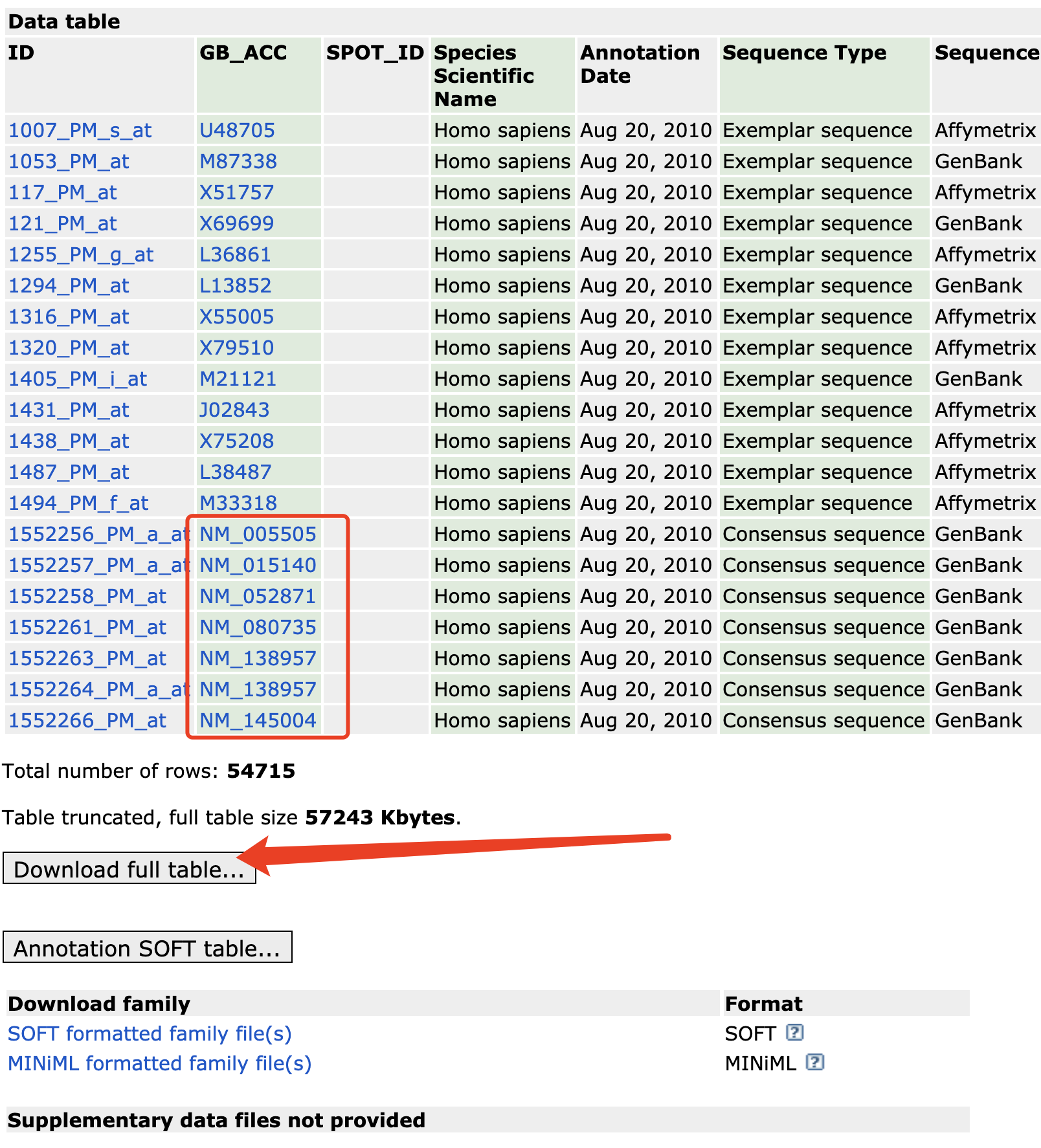
本来呢,我是准备直接回复读者这个 GB_ACC GenBank Accession Number 就是信息所在,但是下载那个约60M的文件 GPL13158-5065.txt ,然后读入R里面,发现其实这个数据集本来就有gene symbol信息了。
Gene Title Title of Gene represented by the probe set.
Gene Symbol A gene symbol, when one is available (from UniGene).
ENTREZ_GENE_ID Entrez Gene Database UID
真的是 超级简单了!
a=read.table('GPL13158-5065.txt',header = T,sep = '\t',fill=T, quote = '')
head(a$Gene.Symbol)
# "DDR1" "RFC2" "NM_002155" "PAX8" "GUCA1A" "UBA7"
可以看到有一些探针对应不到合适的Gene Symbol ,仍然是GB_ACC的refseq的ID,不过应该是没有大问题。
如果要把基因区分成为编码与非编码
有一个包(annoprobe)需要安装,安装annoprobe的代码超级简单:
library(remotes)
# https://git-scm.com/downloads
# 居然有些人电脑里面没有git软件,可怕!!!
url='https://gitee.com/jmzeng/annoprobe.git'
install_git(url)
安装annoprobe的时候不要更新如何其它R包哈 ,但是我看有学生反馈,安装的时候提示他的电脑缺乏git软件,我就很纳闷了,一个搞生物信息学数据分析的电脑里面,怎么可能没有git软件呢?就算是没有,马上去下载git安装也可以啊!!!
也可以自行前往下载gtf文件或者其它方法获取基因的相关信息。
然后简单代码转换即可:
a=read.table('GPL13158-5065.txt',header = T,sep = '\t',fill=T, quote = '')
head(a$Gene.Symbol)
length(unique(a$Gene.Symbol))
library(AnnoProbe)
tmp=annoGene(unique(a$Gene.Symbol),'SYMBOL','human')
tail(sort(table(tmp$biotypes)))
但是有一个警告:
> library(AnnoProbe)
> tmp=annoGene(a$Gene.Symbol,'SYMBOL','human')
Warning message:
In annoGene(a$Gene.Symbol, "SYMBOL", "human") :
37.77% of input IDs are fail to annotate...
> tail(sort(table(tmp$biotypes)))
protein_coding
15509
> length(unique(a$Gene.Symbol))
[1] 20742
我看了看,本来是54732行 的文件,但是一般来说多个探针会对应同一个基因,所以基因数量仍然是2万多个,但是转换的失败率有点高,所以这样的方法仅仅是针对基因名字比较合规的进行了注释。
其实可以直接把所有的 protein_coding 删除,剩下的慢慢看。
pd=tmp[tmp$biotypes =='protein_coding',1]
non=a[!a$Gene.Symbol %in% pd,]
蛮有意思的:
得到的结果如下:
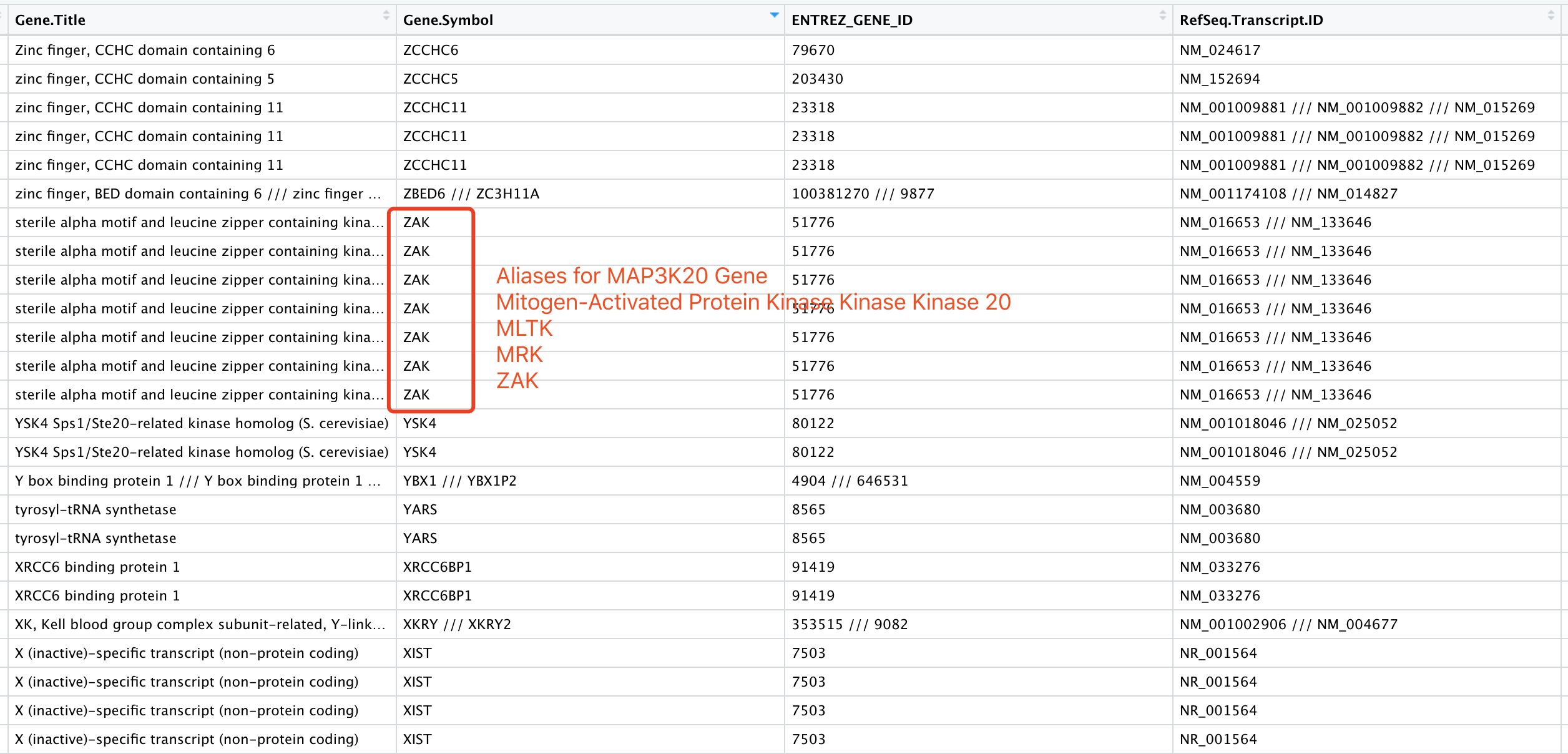
可以看到,这2万多个探针里面,还有四千多个可能是是蛋白编码基因,根据gtf文件是无法成功转换的,因为他们的基因名字都过时了。比较幸运的是,还剩下基因的entrez ID,可以试试看。
library(org.Hs.eg.db)
tmp=AnnotationDbi::select(org.Hs.eg.db,keys = non$ENTREZ_GENE_ID,keytype = 'ENTREZID',columns = c('SYMBOL','ENTREZID'))
tmp=na.omit(tmp)
non=merge(non,tmp,by.x='ENTREZ_GENE_ID',by.y='ENTREZID')
tmp=annoGene(unique( non$SYMBOL ),'SYMBOL','human')
tail(sort(table(tmp$biotypes)))
这样的两个步骤可以查找到一千多个非编码基因。