前面我们提到了因为细胞数量比较多,运行infercnv::run的时候,下面两个参数,都是默认值即可:
HMM参数 when set to True, runs HMM to predict CNV level (default: FALSE)
denoise If True, turns on denoising according to options below (default: FALSE)
这样速度就超级快,可以得到如下图表:

可以看到, 部分Fibroblasts和Endothelial_cells细胞我拿它们作为ref,理论上它们是不可能有CNV事件的,所以上面的热图的上半部分可以看到部分Fibroblasts和Endothelial_cells细胞都是比较整齐。而且,我在需要被检验CNV的细胞里面也掺入了部分Fibroblasts和Endothelial_cells细胞,它们也是没有CNV事件的,仅仅是上皮细胞可以看到泾渭分明的CNV有无的差异。
现在最重要的目标就是根据这个图表或者说inferCNV的结果文件,把上皮细胞里面那些恶性的部分挑选出来,可以跟文章挑选好的进行对比。或者跟我们前面的上皮细胞聚类的结果进行对比!
首先查看inferCNV结果文件夹,可以看到每个步骤的中间文件,都是保存下来了的:
01_incoming_data.infercnv_obj
02_reduced_by_cutoff.infercnv_obj
03_normalized_by_depth.infercnv_obj
04_logtransformed.infercnv_obj
08_remove_ref_avg_from_obs_logFC.infercnv_obj
09_apply_max_centered_expr_threshold.infercnv_obj
10_smoothed_by_chr.infercnv_obj
11_recentered_cells_by_chr.infercnv_obj
12_remove_ref_avg_from_obs_adjust.infercnv_obj
14_invert_log_transform.infercnv_obj
15_no_subclustering.infercnv_obj
但是最重要的文件是:
infercnv.observations_dendrogram.txt
infercnv.observations.txt
解析热图附带的层次聚类结果
主要是该文章的GitHub代码,读取自己走完inferCNV流程后的结果文件夹里面的 infercnv.observations_dendrogram.txt 文件,里面存储着inferCNV的CNV热图的细胞的层次聚类情况:
rm(list=ls())
options(stringsAsFactors = F)
library(phylogram)
library(gridExtra)
library(grid)
require(dendextend)
require(ggthemes)
library(tidyverse)
library(Seurat)
library(infercnv)
library(miscTools)
# Import inferCNV dendrogram
infercnv.dend <- read.dendrogram(file = "plot_out/inferCNV_output2/infercnv.observations_dendrogram.txt")
# Cut tree
infercnv.labels <- cutree(infercnv.dend, k = 6, order_clusters_as_data = FALSE)
table(infercnv.labels)
# Color labels
the_bars <- as.data.frame(tableau_color_pal("Tableau 20")(20)[infercnv.labels])
colnames(the_bars) <- "inferCNV_tree"
the_bars$inferCNV_tree <- as.character(the_bars$inferCNV_tree)
infercnv.dend %>% set("labels",rep("", nobs(infercnv.dend)) ) %>% plot(main="inferCNV dendrogram") %>%
colored_bars(colors = as.data.frame(the_bars), dend = infercnv.dend, sort_by_labels_order = FALSE, add = T, y_scale=100 , y_shift = 0)
如下所示:
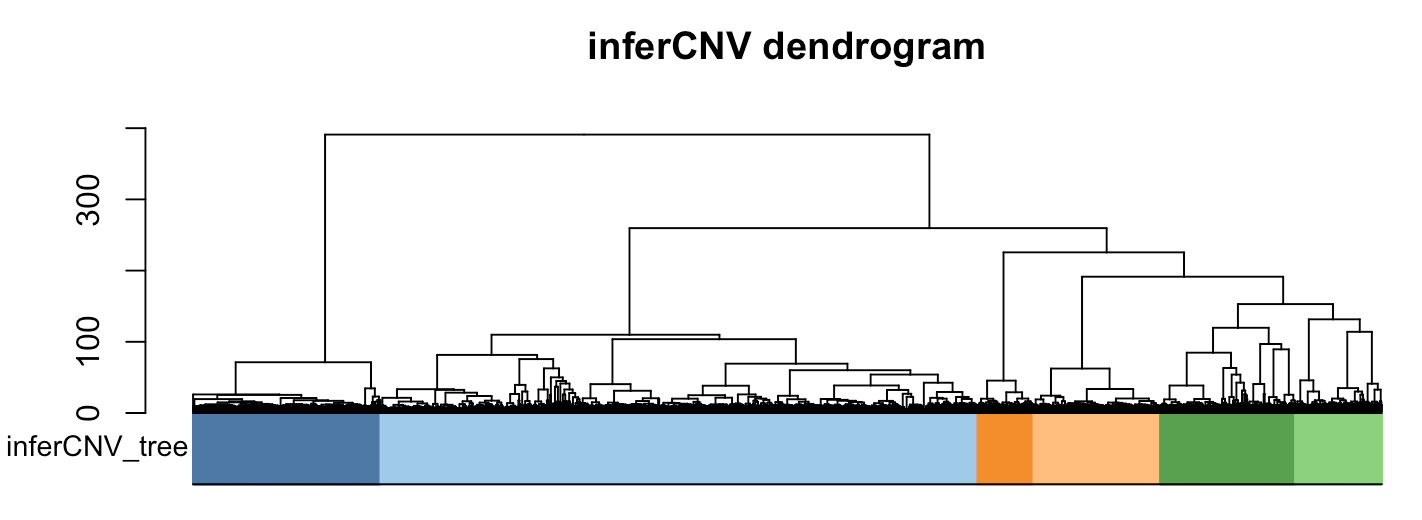
可以很清楚的看到,最大的那一类 就是第二类,有3035个细胞的群体是非恶性细胞,不仅仅是从前面的热图可以看到它们这些细胞基本上没有CNV事件,而且这一群细胞里面有我们自己掺入的已知的非恶性细胞,就是各300个的Fibroblasts和Endothelial_cells细胞。
> table(infercnv.labels)
infercnv.labels
1 2 3 4 5 6
954 3035 285 644 686 440
而且可以仍然是跟之前的spike-in细胞对比:
infercnv.labels=as.data.frame(infercnv.labels)
groupFiles='groupFiles.txt'
meta=read.table(groupFiles)
infercnv.labels$V1=rownames(infercnv.labels)
meta=merge(meta,infercnv.labels,by='V1')
table(meta[,2:3])
得到的结果如下:
> table(meta[,2:3])
infercnv.labels
V2 1 2 3 4 5 6
epi 954 2436 285 644 685 440
spike-endo 0 300 0 0 0 0
spike-fib 0 299 0 0 1 0
自己人为掺入的Fibroblasts和Endothelial_cells细胞都是在第2群,也就是非恶性细胞。而剩余的其它细胞亚群,都是恶性细胞!
根据inferCNV结果判定的细胞恶性与否的结果和普通聚类的差异
在CNS图表复现09—上皮细胞可以区分为恶性与否,我分享过第1,2,7,14,21,23,25 是跨越病人的聚类情况,所以先暂时认为他们是非恶性细胞。现在我们有了inferCNV结果,就可以看看两个策略判断细胞恶性与否的差异:
load(file = 'first_sce.Rdata')
load(file = 'phe-of-first-anno.Rdata')
sce=sce.first
table(phe$immune_annotation)
sce@meta.data=phe
cells.use <- row.names(sce@meta.data)[which(phe$immune_annotation=='epi')]
length(cells.use)
sce <-subset(sce, cells=cells.use)
sce
load(file = 'phe-of-cancer-or-not.Rdata')
sce@meta.data=phe
colnames(sce@meta.data)
table(sce@meta.data$cancer)
sce@meta.data$inferCNV=meta[match(rownames(sce@meta.data),meta$V1),'infercnv.labels']
table(sce@meta.data$cancer,sce@meta.data$inferCNV)
结果如下:
> table(sce@meta.data$cancer,sce@meta.data$inferCNV)
# 其中inferCNV得到的亚群里面,第2群细胞是 非恶性
1 2 3 4 5 6
cancer 948 956 284 640 466 438
non-cancer 6 1480 1 4 219 2
可以看到,通过聚类是否跨越病人来区分恶性与否,跟inferCNV的一致性尚可。
因为之前那个非常重要的文件是: cnv_scores.csv ,在新版的inferCNV里面是没有的,所以我们仅仅是能根据层次聚类的划分情况,来粗略的把第2群的3000多个细胞统一划分成为非恶性。但实际上,很容易看出来:

这个第2群的3000多个细胞是可以继续细分后,重新判定细胞的恶性与否,这样就可以提高两个技术的吻合性。
但是这样仍然是“治标不治本”,无论我们的肉眼多么厉害,仅仅是从这个CNV热图去判断具体的某个层次聚类的亚群细胞是恶性与否,实在是太主观了。
虽然文章作者是靠这样的策略来判断细胞恶性与否
虽然我和文章都是取全部的上皮细胞,以及部分Fibroblasts和Endothelial_cells细胞来一起运行inferCNV流程。上皮细胞肯定都是一样的,但是因为是随机函数取部分Fibroblasts和Endothelial_cells细胞,所以没办法保证我跟文章后面的inferCNV结果一模一样。文章的确可以直接层次聚类后的6类群,就区分出来了恶性细胞,全部代码如下:
#replace inferCNV.class 1 as "tumor"
inferCNV.annotation.malignant$inferCNV.class <- gsub(pattern = 1, replacement = "tumor", x = inferCNV.annotation.malignant$inferCNV.class)
#replace inferCNV.class 2 as "nontumor"
inferCNV.annotation.malignant$inferCNV.class <- gsub(pattern = 2, replacement = "nontumor", x = inferCNV.annotation.malignant$inferCNV.class)
#replace inferCNV.class 3 as "tumor"
inferCNV.annotation.malignant$inferCNV.class <- gsub(pattern = 3, replacement = "tumor", x = inferCNV.annotation.malignant$inferCNV.class)
#replace inferCNV.class 4 as "tumor"
inferCNV.annotation.malignant$inferCNV.class <- gsub(pattern = 4, replacement = "tumor", x = inferCNV.annotation.malignant$inferCNV.class)
#replace inferCNV.class 5 as "tumor"
inferCNV.annotation.malignant$inferCNV.class <- gsub(pattern = 5, replacement = "tumor", x = inferCNV.annotation.malignant$inferCNV.class)
#replace inferCNV.class 6 as "tumor"
inferCNV.annotation.malignant$inferCNV.class <- gsub(pattern = 6, replacement = "tumor", x = inferCNV.annotation.malignant$inferCNV.class)
#update colnames of inferCNV.annotation.malignant
colnames(inferCNV.annotation.malignant) <- c("cell_id", "Epithelial_cluster", "inferCNV_annotation")
table(inferCNV.annotation.malignant$inferCNV_annotation)
但是很明显,这个代码并不适合我自己走一波流程的数据结果解读。我们需要一些量化指标来具体判定每个细胞亚群是否恶性,比如计算具体每个细胞的CNV score。
后面我们来继续分享进阶方案!