很多人问针对RNA-seq数据如何找变异位点,尤其是肿瘤,比如TCGA计划的海量RNA-seq数据,所以我就写了教程:
- 2017年6月:RNA-seq 检测变异之 GATK 最佳实践流程
- 2019年11月:最新版针对RNA-seq数据的GATK找变异流程
并且分享了代码,就是STAR aligner 2-pass的比对,衔接上 GATK的MuTect2流程找变异位点。
最近无意中看到了2018年6月发表在PeerJ的BIOINFORMATICS AND GENOMICS的文章标题是;《Detection and benchmarking of somatic mutations in cancer genomes using RNA-seq data》的文章,我看了看跟我的教程基本上差不多额。
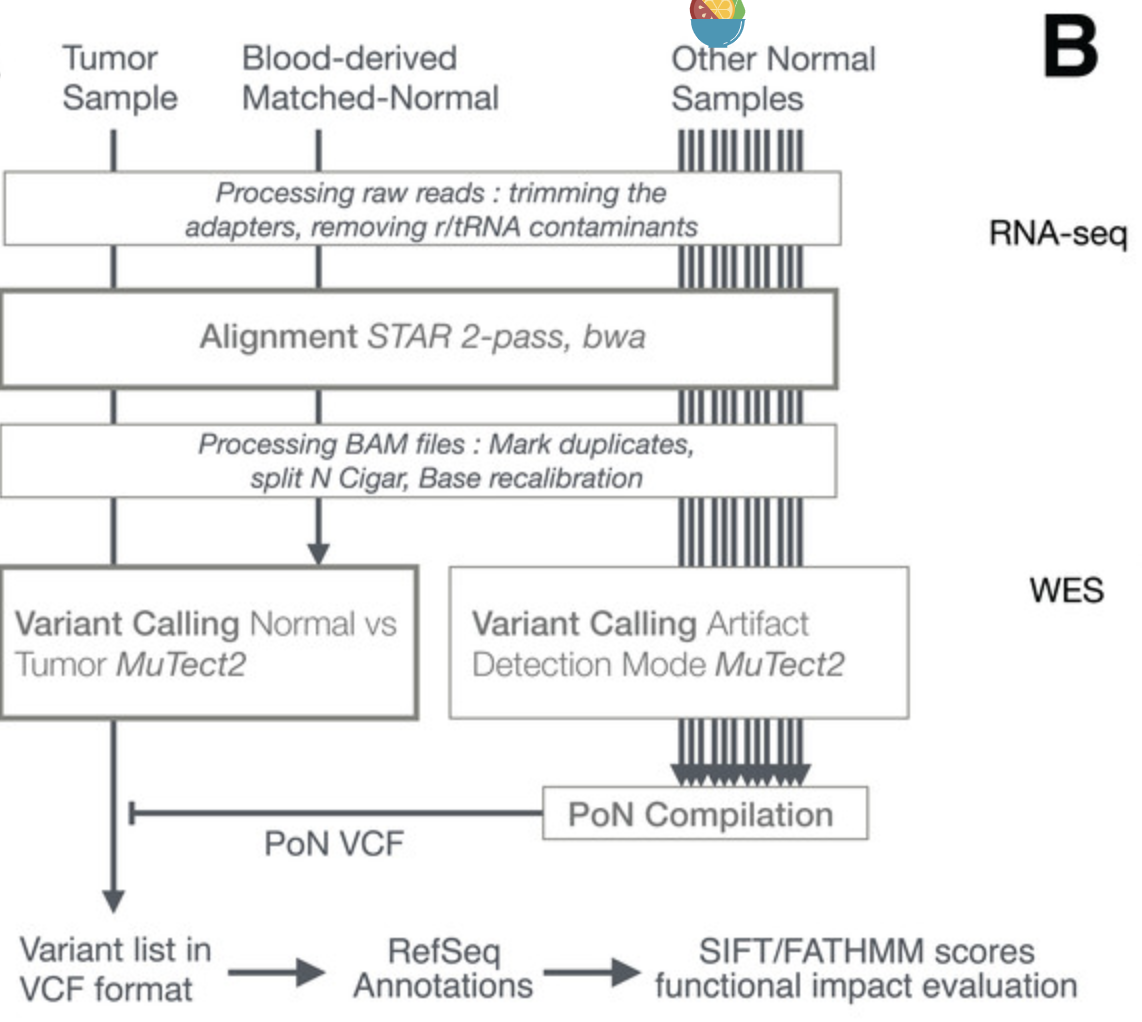
同时拿TCGA数据库的GBM的转录组数据做了测试,生物信息学流程(各个步骤软件选择)如下: - The process first involved trimming the adapters with cutadapt (v1.10) (Martin, 2011) from fastq files,
- removing sequences that were shorter than 36 bases after trimming,
- and removing rRNA and tRNA sequences by aligning with BWA (v0.7.12-r1039) to a reference built with known rRNA/tRNA.
- Filtered reads were then aligned with STAR aligner (v2.4.2a) using a 2-pass procedure (Dobin & Gingeras, 2015).
- Before variant calling, aligned reads in BAM format were sorted, duplicate reads were flagged (MarkDuplicates, Picard v2.5.0),
- the base scores recalibrated (BaseRecalibrator, GATK v3.6)
- and RNA-seq reads were split into exons (SplitNCigarReads, GATK v3.6).
- Variant calling was done with MuTect2 in tumor versus normal mode as described below.
- Variants recovered in VCF files were then separated into RNA-seq-only, Intersection and WES-only.
- ANNOVAR (v.2016Feb01) (Wang, Li & Hakonarson, 2010) was used to annotate variants relative to RefSeq annotations (release 73)
- SIFT score/prediction (v2.3) (Ng & Henikoff, 2003), and FATHMM score/prediction with cancer weights (v2.3) were used to evaluate the functional impact of non-synonymous SNVs and frameshift indels.
流程图如下:
文章链接是:https://peerj.com/articles/5362/ 感兴趣也可以看看,其实如果你比较有空,而且英语比较不错,类似的文章写起来应该是不难的。
不过,其实针对RNA-seq数据的变异,其实也有专门的软件啦,并不需要自己搭建流程,使用STAR aligner 2-pass的比对,衔接上 GATK的MuTect2流程找变异位点。只不过是用得少,不出名。
补充阅读; - https://europepmc.org/article/MED/29091775
- https://rna-seqblog.com/inconsistency-of-somatic-snvs-called-in-wes-and-rna-seq-data/
文末友情推荐
要想真正入门生物信息学建议务必购买全套书籍,一点一滴攻克计算机基础知识,书单在:什么,生信入门全套书籍仅需160 。
如果大家没有时间自行慢慢摸索着学习,可以考虑我们生信技能树官方举办的学习班: - 数据挖掘学习班第7期(线上直播3周,马拉松式陪伴,带你入门),原价4800的数据挖掘全套课程, 疫情期间半价即可抢购。
- 生信爆款入门-第9期(线上直播4周,马拉松式陪伴,带你入门),原价9600的生信入门全套课程,疫情期间3.3折即可抢购。
如果你课题涉及到转录组,欢迎添加一对一客服:详见:你还在花三五万做一个单细胞转录组吗?
号外:生信技能树知识整理实习生招募,长期招募,也可以简单参与软件测评笔记撰写,开启你的分享人生!另外,:绝大部分生信技能树粉丝都没有机会加我微信,已经多次满了5000好友,所以我开通了一个微信好友,前100名添加我,仅需150元即可,3折优惠期机会不容错过哈。我的微信小号二维码在:0元,10小时教学视频直播《跟着百度李彦宏学习肿瘤基因组测序数据分析》