最近安排学员做了一下:Endothelial(单细胞研究调研),见:https://share.mubu.com/doc/6capdKnSkBv

明明Endothelial Cell Marker是 (CD31, CD34, ICAM1, CD45),但是有一个研究却关注了CD105 (Endoglin) ,我就去bing搜索了CD105 (Endoglin) ,发现一个有意思的研究。
发表在Sci Rep. 2019;的文章,标题是:《CD105 (Endoglin) as negative prognostic factor in AML》,本来以为是一个单基因数据挖掘文章, 结果分析他们是自己的 a cohort of 62 AML patients,看CD105的蛋白水平在AML疾病的生存意义。
而且为了拿到统计学显著的生存分析结果,研究者们根据CD105的蛋白水平的quartiles (Q1-4) 分成4组后,主要是看Q1和Q4组的差异。
To correlate CD105 expression in quartiles with OS, Kaplan-Meier analysis was performed and revealed that patients with low or absent CD105 expression (Q1) showed superior survival and did not reach median OS in the observation time, whereas for patients in the highest quartile (Q4) median OS was 284 days (p = 0.0098) (Fig. 3B).
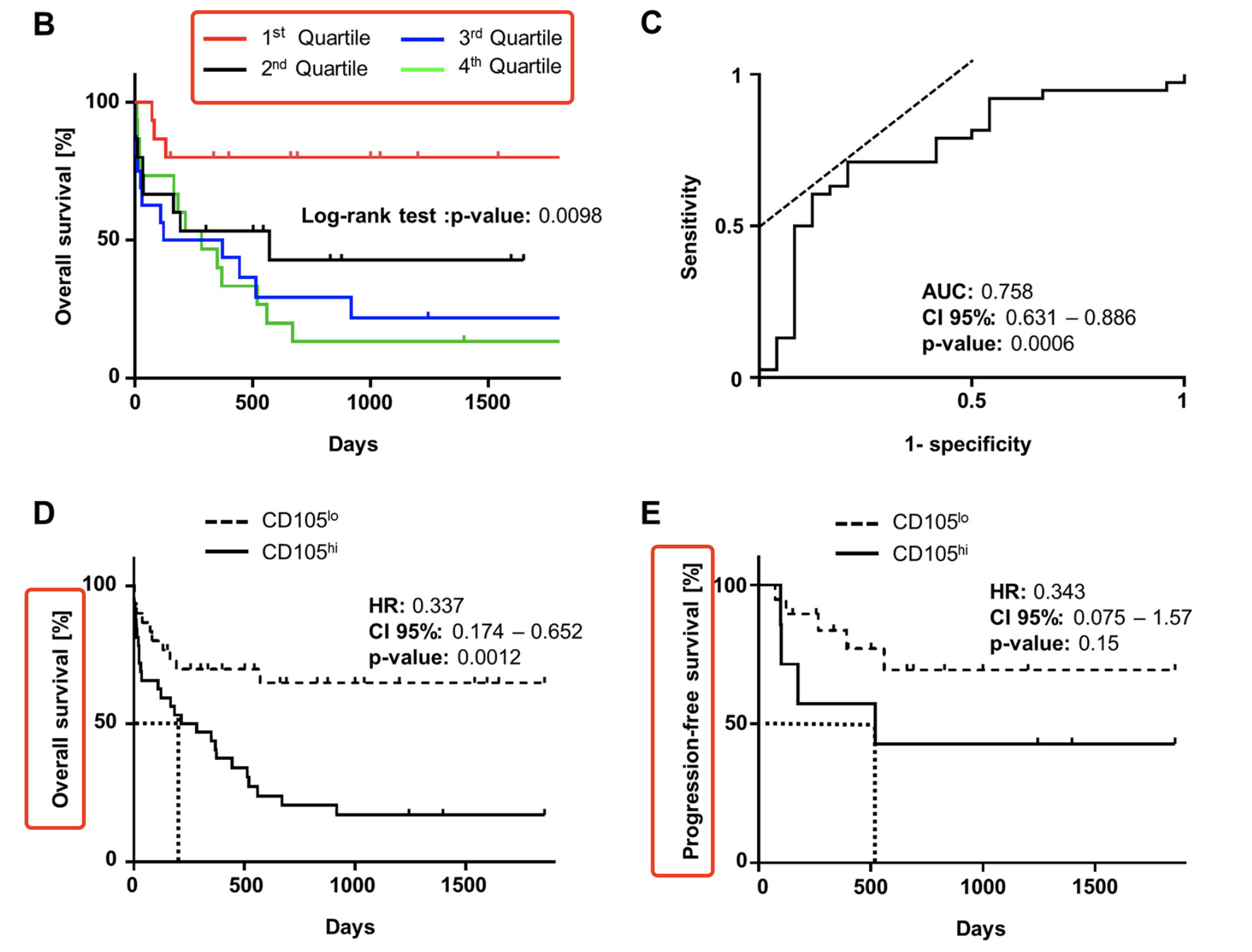
这个生存分析结果很容易解释,如果按照 CD105表达量高低对病人分组呢,没有统计学显著的结果,但是呢,如果把表达量四等分后,再次把病人也对应的分成4个组,针对性的比较表达量最高和最低的两个组,然后就有统计学显著的结果啦!
Next we established a cut-off specific fluorescence level of 5.22 using receiver-operating characteristics, which allowed to group patients in cases with CD105lo and CD105hi surface expression and revealed that high CD105 expression correlated significantly with poor overall and progression free survival.
第一眼看到研究者的这个结论呢,我就想起来了很明显的生存分析网页工具,就可以看TCGA的AML病人队列的该基因的生存预后意义。同样的,在bing搜索了一些TCGA的AML病人队列文章,发表的比较早:
TCGA在N Engl J Med 2013; 的文章,纳入了200 patients,链接:https://www.nejm.org/doi/full/10.1056/NEJMoa1301689
- RNA-expression profiling on the Affymetrix U133 Plus 2 platform for 197 samples,
- RNA sequencing for 179 samples, (一般来说,大家会挖掘这个数据,RNA-seq and mutation data from 176 AML patients from the Cancer Genome Atlas (TCGA) database )
- microRNA (miRNA) sequencing for 194 samples,
- Illumina Infinium HumanMethylation450 BeadChip profiling for 192 samples,
- Affymetrix SNP Array 6.0 for both tumor and normal skin samples
- whole-genome sequencing (50 cases)
- whole-exome sequencing (150 cases)
实际上芯片拿到的表达矩阵和RNA-seq拿到的,是可以都分析一下,相呼应。OncoLnc是最简单的TCGA生存分析网页工具
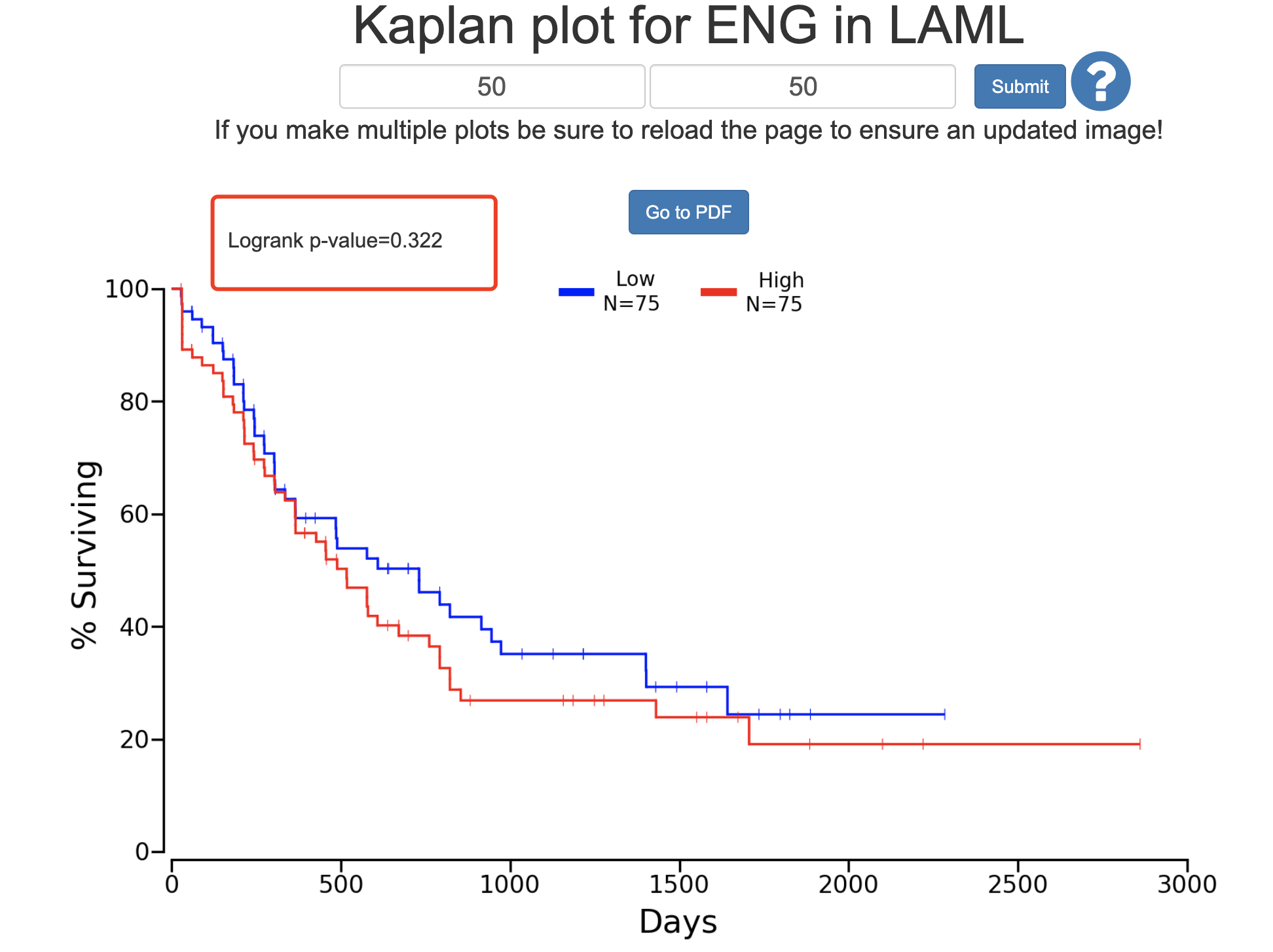
如果我们按照默认参数来进行网页工具查询,可以看到, CD105表达量高低对病人分组呢,没有统计学显著的结果:
- http://www.oncolnc.org/kaplan/?lower=50&upper=50&cancer=LAML&gene_id=2022&raw=ENG&species=mRNA
同样的,我们按照文章的分成4个组,针对性的比较表达量最高和最低的两个组: - http://www.oncolnc.org/kaplan/?lower=25&upper=25&cancer=LAML&gene_id=2022&raw=ENG&species=mRNA
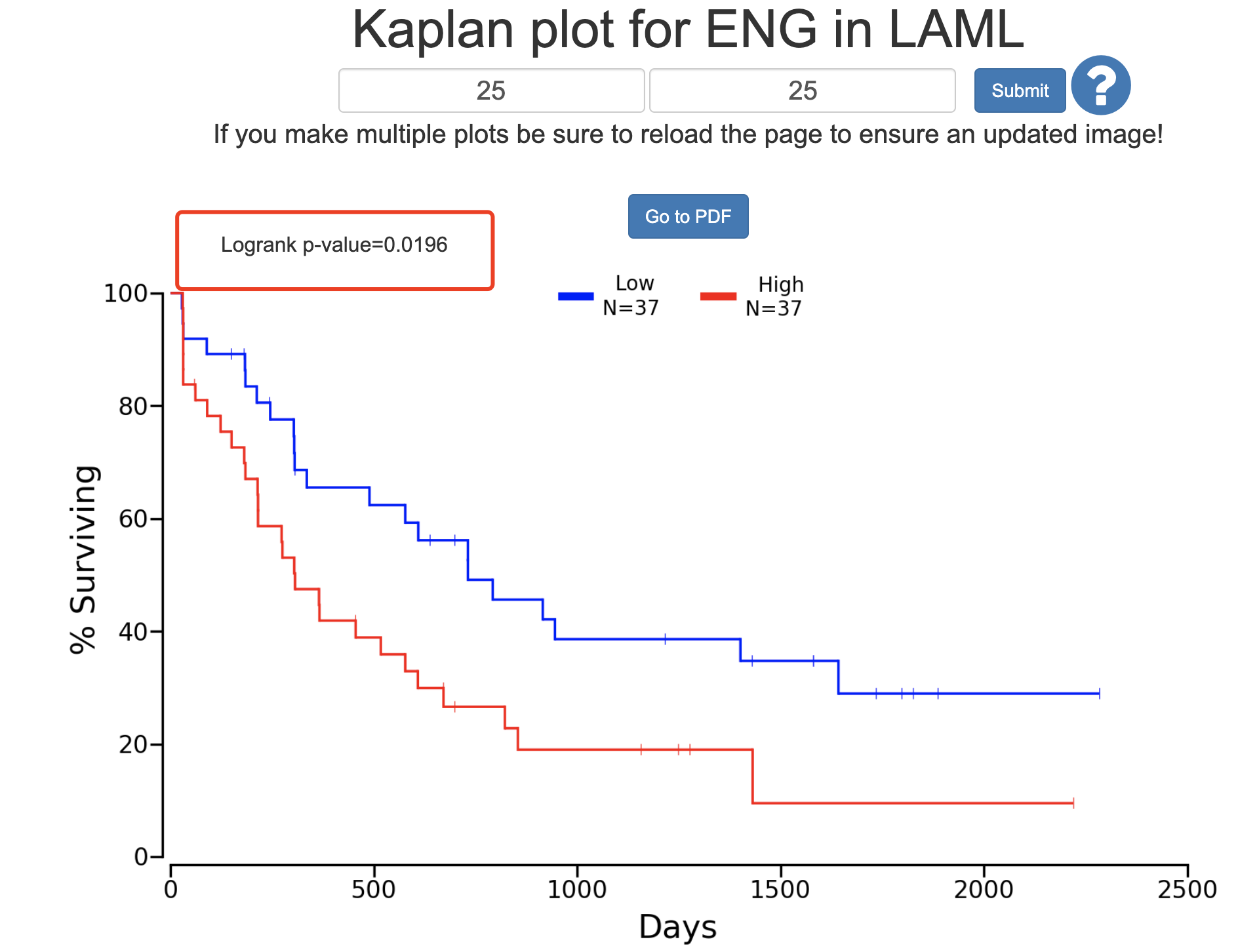
确实达到了统计学显著!
也就是说,这个研究,其实就网页工具同样的结果!公共数据库挖掘这个技能的重要性大家认识不够,其实是能极大程度的避免大家重复浪费科研经费去做一些明明可以通过分析公共数据库拿到的结论!
比如你研究的癌症里面哪些基因高表达,哪些低表达,你通过数据挖掘拿到了感兴趣基因,后续自己设计基础实验来探索它们,完善你的生物学故事。假如你并不知道可以分析公共数据库,那么你就不得不自己去做一次癌症病人队列的转录组,耗费几万块钱来拿到一个本来就可以通过公共数据库分析拿到的上下调基因。或者说,你已经有了比较完整的生物学故事,定位到了具体的通路或者基因,如果想设计病人队列来说明你感兴趣的基因或者通路的临床意义,就是一个大工程,从病人招募信息整理,到ngs组学数据采集,分析,统计可视化等等。但是大概率上你感兴趣的疾病都会有现成的公共数据,你完全可以选择从你感兴趣的角度来对它进行分析。数据挖掘的核心是缩小目标基因
各种数据挖掘文章本质上都是要把目标基因集缩小,比如表达量矩阵通常是2万多个蛋白编码基因,不管是表达芯片还是RNA-seq测序的,采用何种程度的差异分析,最后都还有成百上千个目标基因。如果是临床队列,通常是会跟生存分析进行交集,或者多个数据集差异结果的交集,比如:多个数据集整合神器-RobustRankAggreg包 ,这样的基因集就是100个以内的数量了,但是仍然有缩小的空间,比如lasso等统计学算法,最后搞成10个左右的基因组成signature即可顺利发表。
其实还有另外一个策略方向,有点类似于人工选择啦,通常是可以往热点靠,比如肿瘤免疫,相当于你不需要全部的两万多个基因的表达量矩阵进行后续分析,仅仅是拿着几千个免疫相关基因的表达矩阵即可。
最近比较热门的有:自噬基因,铁死亡,EMT基因,核受体基因家族,代谢基因。还有一个最搞笑的是m6a基因,完全是无厘头的基因集搞小,纯粹是为了搞小而搞小。