最近各个公众号都在鼓吹单细胞转录组数据套路,感觉这样的科研风气不好,数据挖掘这个技能最大的作用应该是避免大家重复浪费科研经费去做一些明明可以通过分析公共数据库拿到的结论!
- 比如你研究的癌症里面哪些基因高表达,哪些低表达,你通过数据挖掘拿到了感兴趣基因,后续自己设计基础实验来探索它们,完善你的生物学故事。假如你并不知道可以分析公共数据库,那么你就不得不自己去做一次癌症病人队列的转录组,耗费几万块钱来拿到一个本来就可以通过公共数据库分析拿到的上下调基因。
- 或者说,你已经有了比较完整的生物学故事,已经定位到了具体的通路或者基因,如果想设计病人队列来说明你感兴趣的基因或者通路的临床意义,就是一个大工程,从病人招募信息整理,到ngs组学数据采集,分析,统计可视化等等。
也就是说,大概率上你感兴趣的疾病都会有现成的公共数据,你完全可以选择从你感兴趣的角度来对它进行分析。而不是跑一下各个标准代码,得到一个唾手可得的结论糊弄大家。科研的科研,教程是教程!
而且我压根就不相信,一两天的填鸭式灌输能让一个完全没有然后变成概念的人掌握R语言并且理解seurat的全部流程,拿到系列分析图表。大家学到的仅仅是类似于网页工具般的鼠标点点点,一键式出图。如下所示:
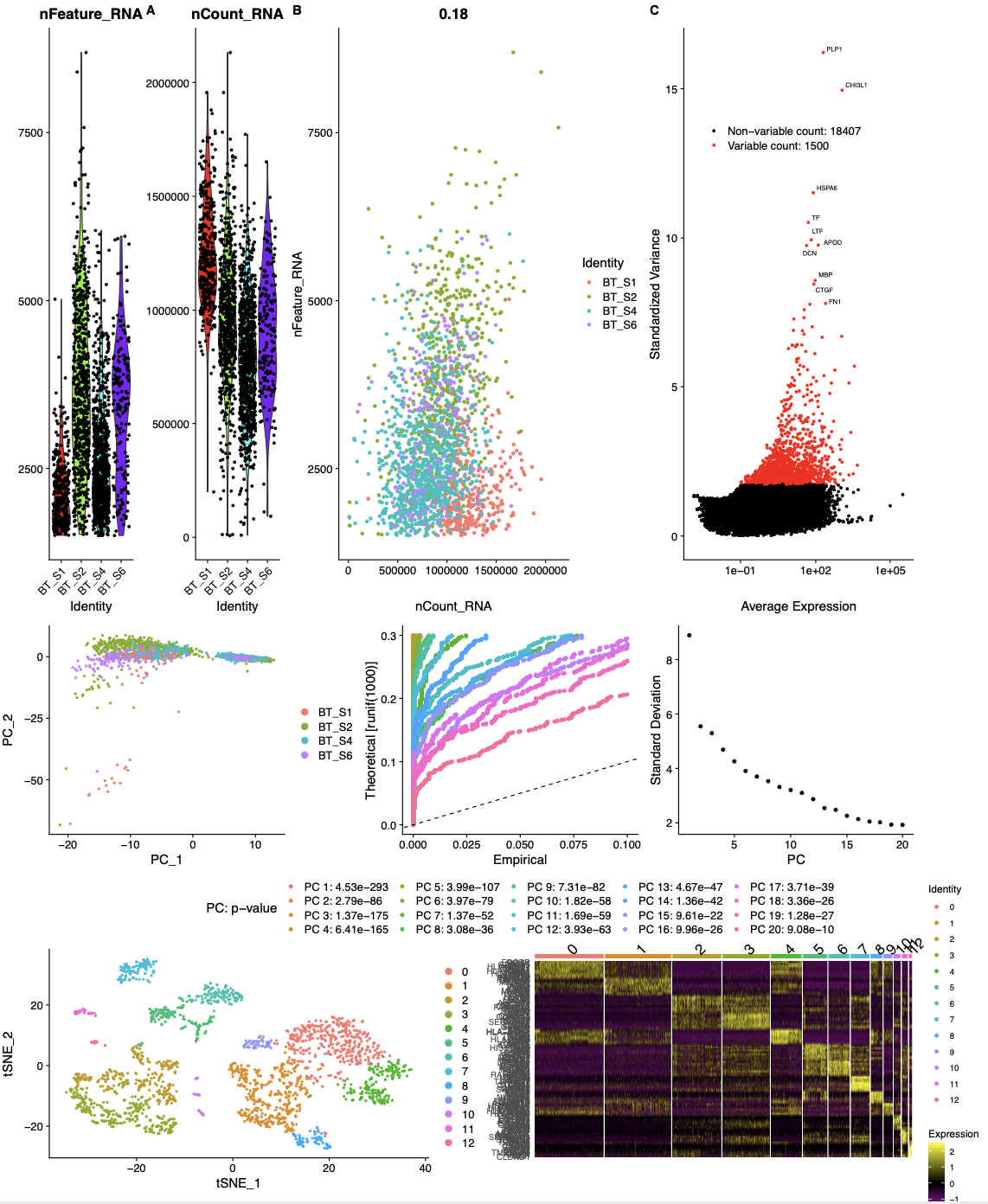
上面的图表基本上就是:跟着seurat学单细胞下游分析 https://satijalab.org/seurat/ 代码原样输出。 (单细胞初识讲义大纲:https://share.mubu.com/doc/3347KRIo18v )

我们《单细胞 天地》团队也多次组织过系列教学活动,最近还有一个CNS图表复现之旅已经开始,前面8讲是;
- 或者说,你已经有了比较完整的生物学故事,已经定位到了具体的通路或者基因,如果想设计病人队列来说明你感兴趣的基因或者通路的临床意义,就是一个大工程,从病人招募信息整理,到ngs组学数据采集,分析,统计可视化等等。
- CNS图表复现01—读入csv文件的表达矩阵构建Seurat对象
- CNS图表复现02—Seurat标准流程之聚类分群
- CNS图表复现03—单细胞区分免疫细胞和肿瘤细胞
- CNS图表复现04—单细胞聚类分群的resolution参数问题
- CNS图表复现05—免疫细胞亚群再分类
- CNS图表复现06-根据CellMarker网站进行人工校验免疫细胞亚群
- CNS图表复现07—原来这篇文章有两个单细胞表达矩阵
- CNS图表复现08—肿瘤单细胞数据第一次分群通用规则
说实话,单细胞转录组可以写教程的已经不多了,但是单细胞转录组本来就是2018-2019年火爆的科研热点,一般来说,一个科研热点就火爆两年,现在单细胞转录组已经演变为了单细胞空间转录组啦。
可是,这次我还没有来得及学习单细胞空间转录组数据分析,就发现了空间单细胞转录组数据挖掘文章。是2020年1月份发表的,文章发的杂志(Breast Cancer Research volume)并不好,但好歹是一个课题,文章题目是:《Identification and transfer of spatial transcriptomics signatures for cancer diagnosis》,链接是:https://pubmed.ncbi.nlm.nih.gov/31931856/ 该研究很容易理解,就是使用机器学习算法来根据单细胞空间转录组数据来区分 non-malignant, DCIS, or IDC,摘要如下:
- Distinguishing ductal carcinoma in situ (DCIS) from invasive ductal carcinoma (IDC) regions in clinical biopsies constitutes a diagnostic challenge.
- We used four publicly available ST breast cancer datasets from breast tissue sections annotated by pathologists as non-malignant, DCIS, or IDC.
- We identified expression signatures for expert annotated regions (non-malignant, DCIS, and IDC) and build machine learning models.
有意思的是,该研究挖掘的那个数据集很出名。是2016年Science文章,利用基因芯片技术将位置信息保留在芯片上,再利用二代测序技术对组织中RNA进行测序,从而生成组织切片上完整的基因表达图像。文章标题是:《Visualization and analysis of gene expression in tissue sections by spatial transcriptomics》,文章链接是:https://science.sciencemag.org/content/353/6294/78
当然了,每年Science文章那么多,如果仅仅是发个Science,不过泯然众人矣。那为什么说这个2016的science文章出名呢。因为2018年底,10X Genomics宣布收购Spatial Transcriptomics,并于2019年11月发布Visium空间基因表达解决方案(Visium Spatial Gene Expression Solution)。而2016年Science文章的通讯作者之一就是瑞典皇家理工学院Joakim Lundeberg(瑞典Spatial Transcriptomics公司联合创始人)。
值得一提的是,另外一个空间单细胞技术就没有那么好的运气了,只能是发个CELL文,可能是因为navin的这个研究专注于基因组测序吧:topographic single cell sequencing(TSCS),以比较原位导管癌(DCIS)区域和浸润性导管癌(IDC)区域细胞之间基因组变异。
- 将组织切片放在载玻片上,用苏木精和伊红(H&E)染色,以便在显微镜下识别单个细胞核。
- 然后使用LCM系统从组织切片中捕捉单个细胞,同时记录每组坐标。
- 接下来,将细胞分离到一个预先编码的缓冲液中,在缓冲液中进行裂解和全基因组扩增(WGA)。
- 最后,对所有cDNA的NGS建库测序。通过LCM总共分离到1293个单细胞,随后进行单核测序(SNS)。
详见我2018年对它的解读 空间单细胞DNA测序:发表于2018年CELL杂志,文章题目是:Multiclonal Invasion in Breast Tumors Identified by Topographic Single Cell Sequencing