作者 so_zy, 2020-10-14
写此文档的缘由:在做GSEA分析时,由于研究的是非模式生物,从Broad Institue开发的MSigDB没有找到合适的预设基因集,没办法顺利进行GSEA. 但是KEGG数据库收录有目标物种。几经折腾,终于跑上了GSEA. 写此文档为其他研究非模式生物的人员提供一点借鉴。
以大熊猫为例:
1. 安装并加载R包
正常情况下,大家安装R包应该是都问题不大了。
#清空当前变量
rm(list = ls())
options(stringsAsFactors = F)
#设置镜像
options("repos"= c(CRAN="https://mirrors.tuna.tsinghua.edu.cn/CRAN/"))
options(BioC_mirror="http://mirrors.ustc.edu.cn/bioc/")
#BiocManager安装"KEGGREST",
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("KEGGREST")
#加载"KEGGREST"
library(KEGGREST) #用于提取通路及基因信息
#查看KEGGREST说明书
browseVignettes("KEGGREST")
#加载clusterProfile
library(clusterProfile)#用于GSEA富集分析
#加载stringr,用于字符串处理
if(!require(stringr))install.packages('stringr')
library(stringr)
2.查询大熊猫在KEGG数据库中的缩写
#获取KEGG数据库收录的所有物种的清单
org <- keggList('organism')
# 在中国大陆地区耗时2-3分钟,在海外耗时一秒钟不到。
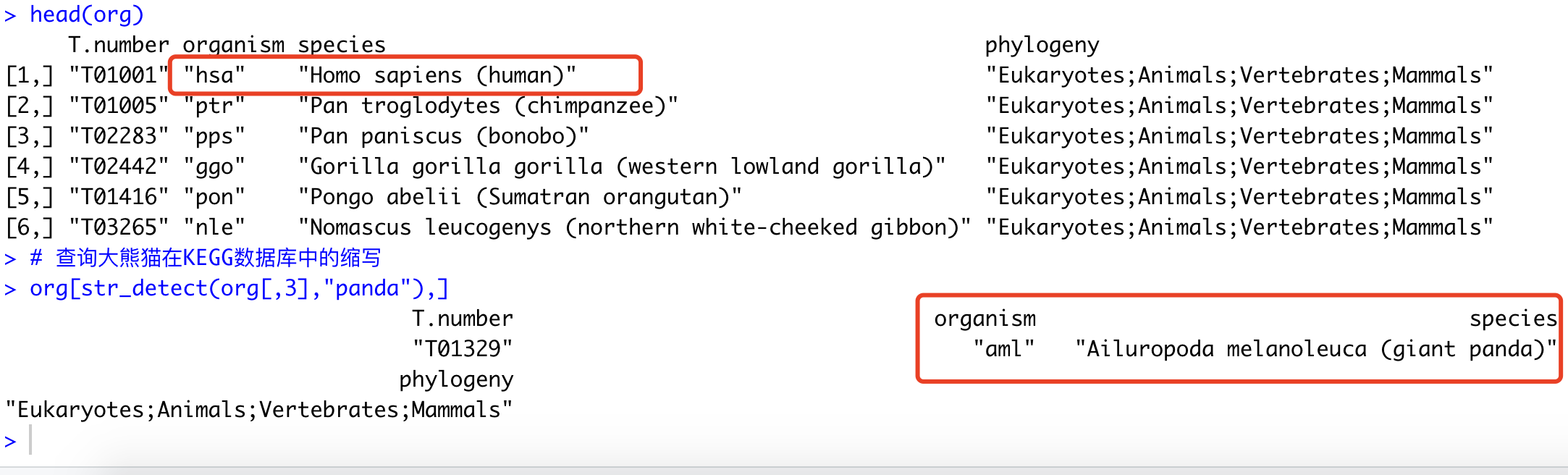
head(org)
# 查询大熊猫在KEGG数据库中的缩写
org[str_detect(org[,3],"panda"),]
当然,也可以网页查询。https://www.genome.jp/kegg/catalog/org_list.html
可以看到,大熊猫在KEGG数据库对应的缩写为“aml”
最出名的物种当然是人类了,人类数据分析超级便捷,到处是造好的轮子。
3.获取大熊猫的KEGG通路及基因集
aml_path <- keggLink("pathway","aml")
#得到字符型向量。元素名为基因id,元素为通路名. 耗时4-5分钟
#查看aml_path的前6个
length(aml_path)
aml_path[1:6]
length(unique(names(aml_path)))
length(unique(aml_path))
可以看到大熊猫的KEGG通路有333条,涉及到的基因数量是7893个(2020-10-14 查询),跟人类研究不相上下哦。
4.获取用于GSEA的基因集数据框
#数据整理,将向量转变为数据框,作为GSEA的基因集
aml.kegg <- data.frame(term=unname(aml_path),gene=names(aml_path))
#将"gene"列中的“aml:”删掉
aml.kegg$gene <- str_replace_all(aml.kegg$gene,"aml:",'')
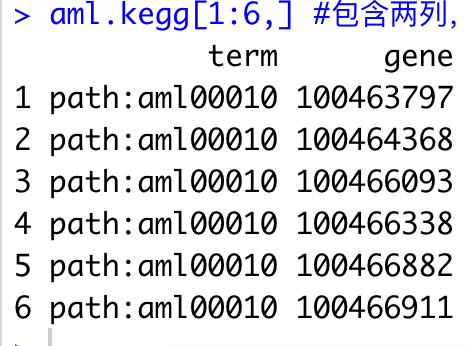
aml.kegg[1:6,] #包含两列,一列term为通路名称,一列gene为基因id
如下所示,基本的数据整理能力:
5.利用clusterProfile进行GSEA
(前提是已获得排序好的genelist)
genesets <- aml.kegg
# 其中这个 genelist 来源于自己的大熊猫转录组数据分析后的基因排序的向量哦。
#富集分析
egmt<- GSEA(genelist, TERM2GENE=genesets, verbose=T,pvalueCutoff = 1)
library(clusterProfiler)
#提取富集结果
kegg_gsea_panda <- as.data.frame(egmt@result)
colnames(kegg_gsea_panda)
#保存结果到当前工作目录
write.table(kegg_gsea_panda,"kegg_gsea_panda.xls",row.names = F,
sep="\t",quote = F)
PS: genelist和genesets都用的是gene ID, 因此这里直接用gene ID进行mapping. 没有将ID转换为symbol.
参考网址:
感谢生信技能树的《生信入门课程转录组讲师》张娟老师的帮助!