在数据挖掘课程研发的早期(2020开春,疫情期间),我们就有预感,它会成为医学生/临床医师首选技能提高课,诚意满满的3周马拉松式授课,第一期就收到了非常棒的评价,见:数据挖掘第一期学习反馈,一个季度过去了,第四期数据挖掘课程也顺利落幕,同样的有学员乐于跟大家分享她参加学习班的笔记和心得体会,如果你看完后觉得有必要,可以考虑一下我们生信技能树举办的最适合医学生/临床医师的数据挖掘课程,8月3号新的一期起航,希望有你!招生宣传见:数据挖掘学习班第5期(线上直播3周,马拉松式陪伴,带你入门),原价4800的数据挖掘全套课程, 疫情期间半价即可抢购。
1.R与Rstudio
主要是学习到了会创建project啊,之前不会如此高效整理自己的项目….都是直接复制粘贴代码进去,所以各种报错,唉。
然后自从开了别人的一个文件project之后,然后回不到最开始的界面了,也不知道之前在哪个文件夹…setwd()都不知道set去哪里
嗯嗯嗯?竟然光标放在那一行的任意位置都行…我蠢啊,虽然没什么区别,但是就像是知道了了不起的小技巧一样。快捷键是ctrl+enter
2.数据类型
确实,学习的过程中不管学习到什么,肯定的说出自己的结论,错了也还有改正的机会呀!
Alt 加 - 可以直接得到 <-,但是一般情况下其实还是可以用=代替。
额我突然想起来在某一次代码出问题时,改过这个数据框的名字问题,明天在茫茫数据中找找,可能还比较显眼,应该是红色的错误。
match的知识点在7-7-3数据结构介绍的17min
7-9-2可以再看几遍。
3.数据结构
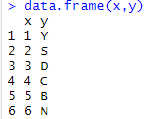
两个不同的向量可以用cbind组合为矩阵,但是矩阵的长度相同,数据类型相同,所以下面x,y是不同的数据类型,是数据框咯?
x=c(1,2,3,4,5,6)
y=c("Y","S","D","C","B","N")
cbind(x,y)

(PS: 学员的推理是错的,虽然最开始,x和y确实数据类型不一样,理论上不可能存在一个矩阵里面,但是呢,用cbind起来后,x和y就都成为了字符,所以可以存在一个矩阵了。)
x=c(1,2,3,4,5,6)
y=c("Y","S","D","C","B","N")
data.frame(x,y)
如果打函数时,遇到比较长需要自动补齐的函数,在出现选项时,可以直接上下键来挑选,然后点Tab或者enter键自动补齐
老师推荐了一个everything的软件,找东西比电脑内带的快很多。
4.函数和R包
安装R包可能会出现”is not available (for R version 3.5.2)“
主要有下面三个原因:
- 1.包名写错
- 2.安装命令使用错误
- 本机的R语言版本与包所要求的版本不符(极少)
browseVignettes(“ggplot2”) 如果不知道某个包的使用方法,可以直接给出这样的代码来查看使用方法和示例,当然也是可以用?ggplot2和example(ggplot2)5.进阶知识
如果有什么包装不上,什么空间原因,可能是包的版本更新了,直接去原始的包存放的地方删掉这个包,重新装就好了。
GEO挖掘流程里,在之前我自己通过东抄一点西抄一点的组成的完整的一篇代码,总觉得自己的分组信息可能出了点问题,但是经过自己多次验证和GEO2R验证以及这次跟小洁老师的代码做对比,发现结果还是没有问题的,让我对自己的数据分析更有信心了。8.下游分析常见图表介绍
之前我以为我了解的很清楚了的地方现在一听原理,也许不是那么回事,但是更清晰明了了。
P.value经常性等于这种格式3.495e-12,后面的12竟然表示的是10的多少次方,厉害鸭。我居然连科学计数法都不知道,果然每个人都有自己的知识盲区。

然后讲了一个新的思路,就是可以取两个不同的疾病联合在一起取差异基因,
热图聚类样本和正常没有聚类到一起,可以画图的矩阵标准化一下?什么意思7-18-1 6.45rm(list = ls()) options(stringsAsFactors = F) library(tinyarray) library(stringr) gse = "GSE42872" geo = geo_download(gse) group_list = ifelse(str_detect(geo$pd$title,"Control"),"control","treat") group_list = factor(group_list,levels = c("control","treat")) find_anno(geo$gpl,install = T) ids <- toTable(hugene10sttranscriptclusterSYMBOL) try = get_deg_all(geo$exp, group_list, ids, logFC_cutoff = 1, entriz = F, adjust = F,heat_id = 1 ) try$plots en = quick_enrich(try$cgs$deg$diff$diffgenes) en[[4]] names(en)然后小洁老师因为讲课时学员上课氛围良好,而主动增加了授课时长。讲解了ceRNA网路的数据挖掘,让我受益匪浅,我也想试着重复一下其它文章的图表。
自己随便找一篇文章复现图。
文章标题是:Comprehensive Analysis of Competing Endogenous RNA (ceRNA) Network Based on RNAs Differentially Expressed in Lung Adenocarcinoma Using The Cancer Genome Atlas (TCGA) Database
首先是下载数据
刚开始想用简便一点的办法,用TCGAbiolinks下载数据,飞快,但是卡在了expda这一步,重复了几次,dyplr也重新安装了,但是问题无法解决,一次运行超慢。遂用了复杂的办法,直接用gdc client下载,听课的时候没有跟着运行,自己跑起来一言难尽,也算是基本掌握了这个下载方式吧,其实也还行,就是下载数据比起上一个工具来,蜗牛一样慢。
: Warning messages: 1:select_()is deprecated as of dplyr 0.7.0. Please useselect()instead. This warning is displayed once every 8 hours. Calllifecycle::last_warnings()to see where this warning was generated.
我i还是不懂这个,明明没有选择miRNA数据,为什么作者说下载了594个样本,然后包含mRNA, lncRNA, and miRNA expression data?他用在线数据库做了CeRNA网络分析,这一点我又得去看下回放了,第一遍基本没起到什么作用,done. - 下载完了数据,使用与作者一样的edgeR包取了差异基因,崩了,完全不一样,他“筛选了2551个差异表达的mRNA,包括2033个上调和518个下调,1359个差异表达的lncRNA,包括1185个上调和174个下调,99个差异表达的miRNA,包括78个上调和21个下调。”而我总共才3833个上调,772个下调(我猜测可能是因为上一步我取了594个样本数据的75%,而作者使用TCGA biolinks后并没有取这个值),不管了,继续往下,我还没有分类。
- 下面就是看看肿瘤样本和正常样本的差别怎么样,这好像有点差
- 差异基因后热图
- 热图和火山图拼到一起(画这个图卡死好几次,垃圾电脑口吐芬芳)
- 生存分析(年龄中竟然有好多缺失值?不过不是用年龄来分析,应该不要紧)
- 不,年龄中有缺失值还是影响了。那是1.删掉有缺失值的样本还是2.使缺失值在分组中也是缺失值呢?目前我两种方法好像都不会欸,找一找。
先是使用这个 meta3 = meta2[na.omit(meta2$age),]但是结果是少了30行确实数量是对的,但是并没有删除缺失值的行啊,删除了个什么?table(is.na(meta2$age)) meta2 = meta2[complete.cases(meta2$age),]ok,成功
- 生存分析出图
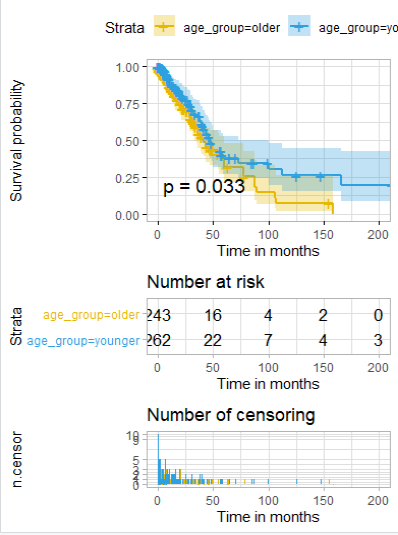
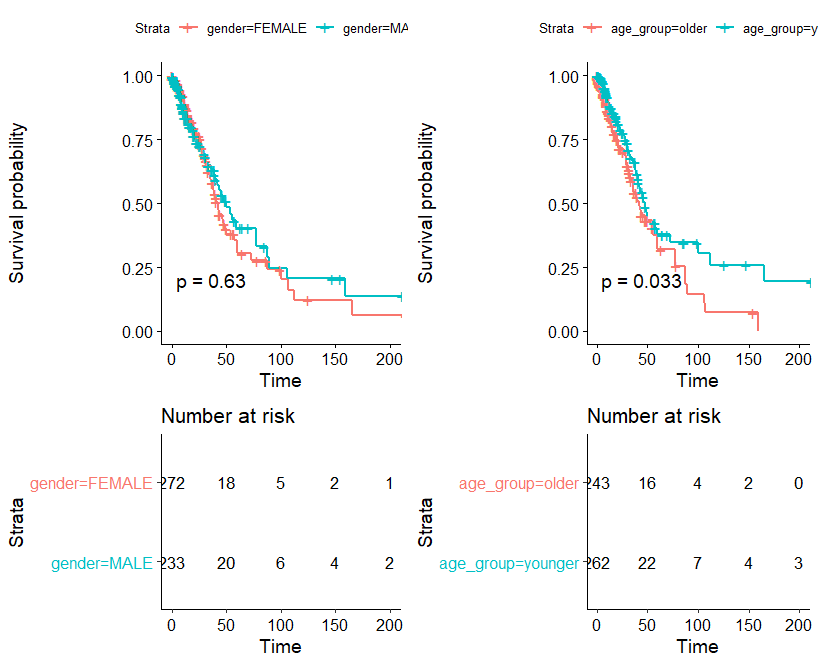
-
- 行吧,因为前面删除了30个数据,后面又出了问题,exprSet还是535列,但是这就要涉及到exprSet的列名的前12个和meta的行名要对应起来,并且寻找出我之前删掉了哪些确实的不匹配的….饶了我吧,我已经预感到很多东西都出了问题,但是如果不删除缺失值,那么年龄分组缺失值不就没有组?难道还单独分出一个组放缺失值吗?也不行。
但是这个匹配又删除怎么搞…..
```
exprSet1 = exprSet
colnames(exprSet1)
rownames(meta)
- 行吧,因为前面删除了30个数据,后面又出了问题,exprSet还是535列,但是这就要涉及到exprSet的列名的前12个和meta的行名要对应起来,并且寻找出我之前删掉了哪些确实的不匹配的….饶了我吧,我已经预感到很多东西都出了问题,但是如果不删除缺失值,那么年龄分组缺失值不就没有组?难道还单独分出一个组放缺失值吗?也不行。
library(stringr)
tmp = str_split(colnames(exprSet1),’-01A’,simplify = T)[,1]
str_count(tmp,”TCGA”)
str_locate(tmp,”TCGA”)[,1]
table(tmp)
length(tmp)
colnames(exprSet1) = tmp
exprSet2 = exprSet1
这样为什么不行?删除了25个是什么
exprSet2 = exprSet2[,rownames(meta) %in% colnames(exprSet2)==”TRUE”]
exprSet2 = exprSet2[,colnames(exprSet2) %in% colnames(meta1)]
table(rownames(meta) %in% colnames(exprSet2))
```
?最后发现不管怎样删除都不行了,数量为什么会不对等呢。
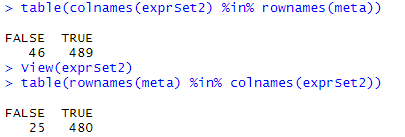
- 然后我直接返回前面去没有删掉缺失值不取年龄分组,然后继续做下去,发现也得不到后面的图,卒。

- 还需要进一步研究
小总结
首先我特别感谢Jimmy大大给了我这次跟着学习的机会,如果不是鼓起勇气发了一次邮件,可能我现在还在咸鱼,每天就打打酱油的度过研究生生涯。现在最起码有了一个清晰的目标,每天知道自己在干什么,应该干什么。抱紧大腿!!!
- 最大的感受:以前每天学习到了一点知识就觉得满足了,美其名曰每天进步一点点,现在就是“这哪够啊,一天时间太短了”。
- 直观的体现:手机每天定时了6小时,以前每天超时,但是最近自打跟着Jimmy大大以来,到晚上还能剩下3小时,这都差不多能戒掉手机,从脑子里装满垃圾娱乐信息到知识??
其次小洁老师太棒了,虽然很多时候听的云里雾里,但是重看一遍,加自己运行一下代码,其实一切都还ok,多看几遍就会觉得清晰明了,老师功底太强了,坑都提前说出来了,但是如果不掉进坑里,还是听不懂为什么这里有个坑哈~简直是速成
确实得多看几遍!听这个数据挖掘的课跟下来还不错,代码运行不下去需要修改的地方在其他的流程中也能找到,只需要看懂代码,稍微改一下也是能改出来的。充实的一周,由完全不懂这个,到差不多能看懂,速成TCGA,太激动了需要多加练习。
文末友情推荐
要想真正入门生物信息学建议务必购买全套书籍,一点一滴攻克计算机基础知识,书单在:什么,生信入门全套书籍仅需160 。
如果大家没有时间自行慢慢摸索着学习,可以考虑我们生信技能树官方举办的学习班: - 数据挖掘学习班第5期(线上直播3周,马拉松式陪伴,带你入门),原价4800的数据挖掘全套课程, 疫情期间半价即可抢购。
- 生信爆款入门-第7期(线上直播4周,马拉松式陪伴,带你入门),原价9600的生信入门全套课程,疫情期间3.3折即可抢购。
如果你课题涉及到转录组,欢迎添加一对一客服:详见:你还在花三五万做一个单细胞转录组吗?
号外:生信技能树知识整理实习生招募,长期招募,也可以简单参与软件测评笔记撰写,开启你的分享人生!另外,:绝大部分生信技能树粉丝都没有机会加我微信,已经多次满了5000好友,所以我开通了一个微信好友,前100名添加我,仅需150元即可,3折优惠期机会不容错过哈。我的微信小号二维码在:0元,10小时教学视频直播《跟着百度李彦宏学习肿瘤基因组测序数据分析》