RobustRankAggreg包在各种数据挖掘文章里面亮相的频次之高,无需我多言,大家可以去查看一下引用它的文献,基本上都是GEO数据库挖掘文章:
比如发表在peerJ的BIOINFORMATICS AND GENOMICS的文章Identification of key candidate genes and biological pathways in bladder cancer 里面的:
作者把这4个数据集,分别独立走差异分析,火山图,热图等等标准流程,基本上读一下我在生信技能树的表达芯片的公共数据库挖掘系列推文就明白了;
- 解读GEO数据存放规律及下载,一文就够
- 解读SRA数据库规律一文就够
- 从GEO数据库下载得到表达矩阵 一文就够
- GSEA分析一文就够(单机版+R语言版)
- 根据分组信息做差异分析- 这个一文不够的
- 差异分析得到的结果注释一文就够
你也可以很轻松的分析这几个数据集: GSE7476, GSE13507, GSE37815 and GSE65635 ,然后作者就使用了RobustRankAggreg包对这4个数据集的差异分析结果进行整合,如下:
- The integrated DEGs were screened using the RRA package (corrected P < 0.05, logFC > 1 or −logFC < −1).
- The RRA method is based on the assumption that each gene in each dataset is randomly arranged.
- If the gene ranks high in all datasets, the associated P-value is lower, the possibility of differential gene expression is greater.
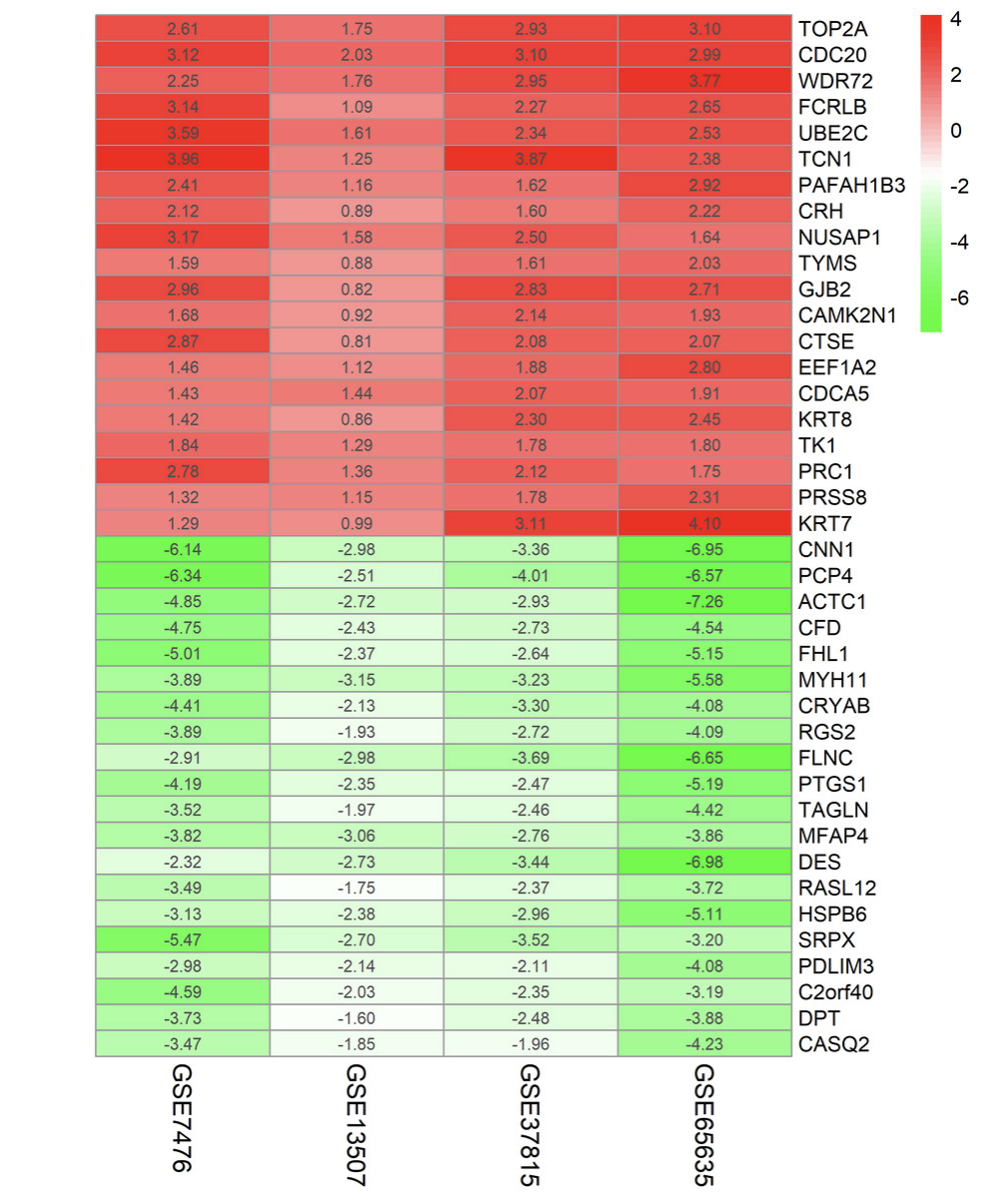
- Through rank analysis, 343 integrated DEGs, consisting of 111 upregulated genes and 232 downregulated genes, were identified by the RRA method
并且把top20的上调基因和下调基因的差异倍数进行热图可视化,如下:
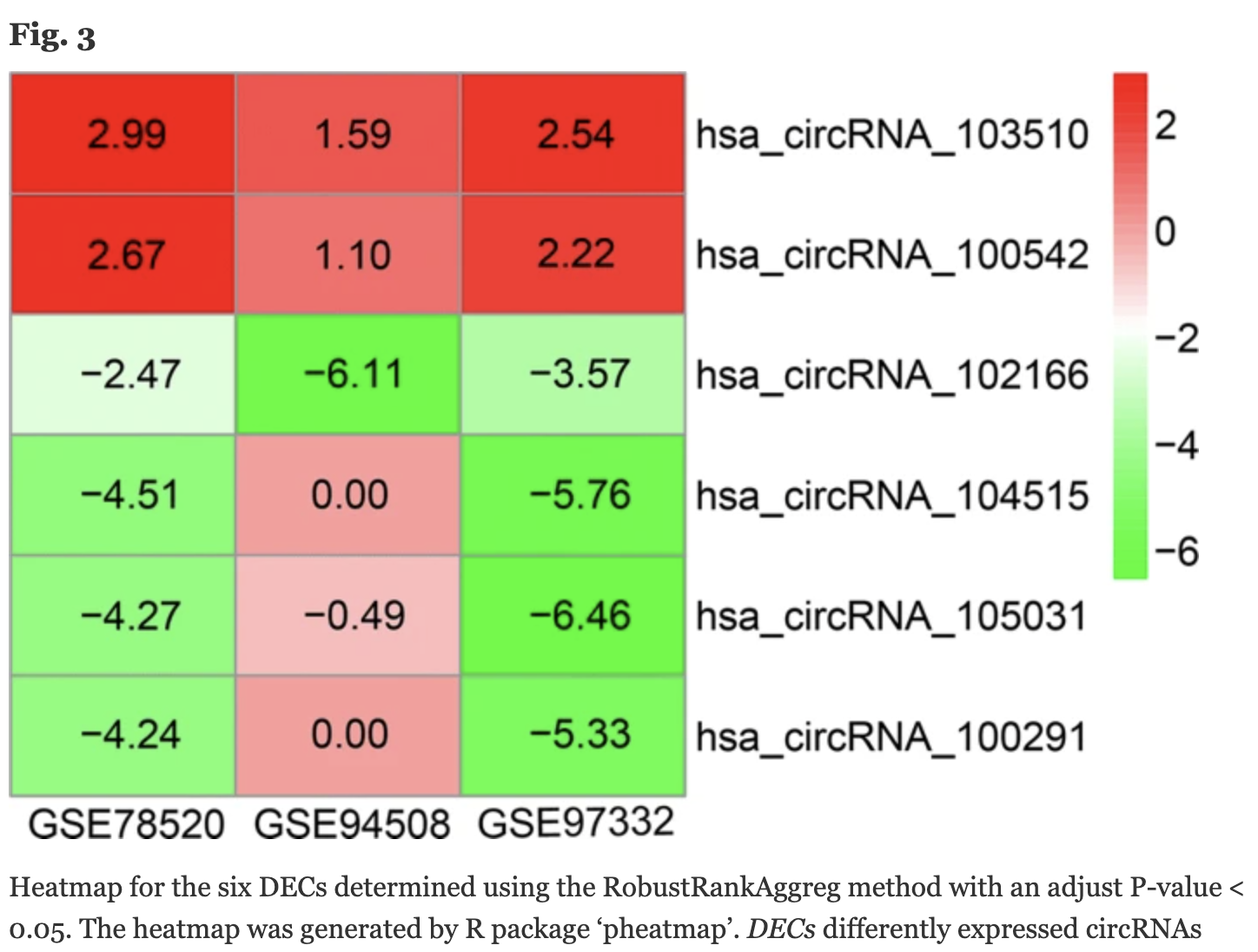
当然了,不仅仅是mRNA的表达芯片,其它,比如circRNA芯片也是如此,同样是发表于2018的文章:A circRNA–miRNA–mRNA network identification for exploring underlying pathogenesis and therapy strategy of hepatocellular carcinoma
就是下载了3个GEO数据集,走差异分析,并且使用RobustRankAggreg包进行整合,最后仅仅是确定了6个circRNA。
几百篇文章我们就不用一一解读啦,反正都是独立的数据集自己做自己的差异分析,然后把多个数据集的差异基因拿去使用RobustRankAggreg包进行整合。
RobustRankAggreg包说明书
这个RobustRankAggreg包超级简单,有意思的是居然并不在bioconductor列表哦,可能是因为它最开始并不是为生物信息学领域的数据分析而创造的吧!因为不在bioconductor,所以它的示例教程一塌糊涂,需要一点背景才能理解。其重点就是aggregateRanks函数而已:
options(BioC_mirror="https://mirrors.tuna.tsinghua.edu.cn/bioconductor/")
options("repos" = c(CRAN="https://mirrors.tuna.tsinghua.edu.cn/CRAN/"))
# https://bioconductor.org/packages/release/bioc/html/GEOquery.html
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("RobustRankAggreg",ask = F,update = F)
library(RobustRankAggreg)
?aggregateRanks
一般来说,正常R包的函数,都是可以通过问号来调取其帮助文档的,aggregateRanks函数也不例外。我们直接看一下示例代码:
set.seed(1234567)
glist <- list(sample(letters, 4), sample(letters, 10), sample(letters, 12))
freq=as.data.frame(table(unlist(glist)))
# Aggregate the inputs
ag=aggregateRanks(glist = glist, N = length(letters))
ag$Freq=freq[match(ag$Name,freq$Var1),2]
的确是超级简单,可以看到,我们有26个字母,假设是26个基因,然后做了3次随机抽样,假设是3个数据集的差异分析,拿到的上调基因,列表如下:
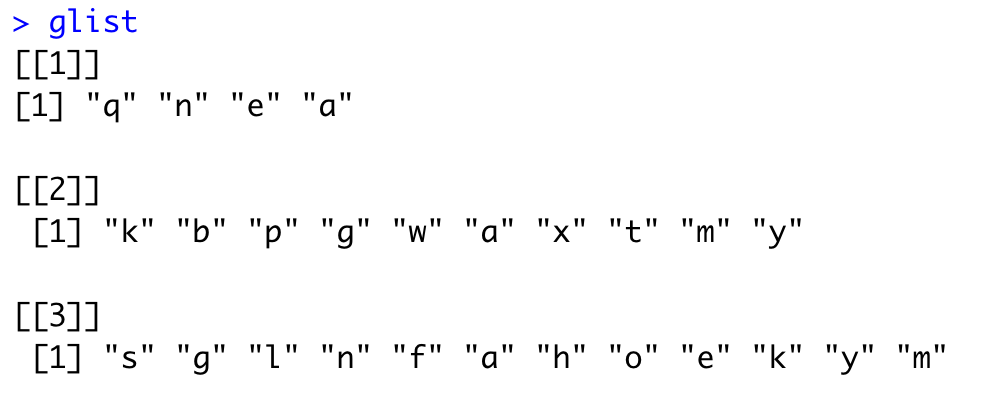
值得注意的是,每次抽样,得到的字母列表的顺序也是有意义的哦。我们的多次数据集差异分析结果,也制作成为这样的表格即可哈!
然后直接使用aggregateRanks函数即可,得到的数据结果如下:
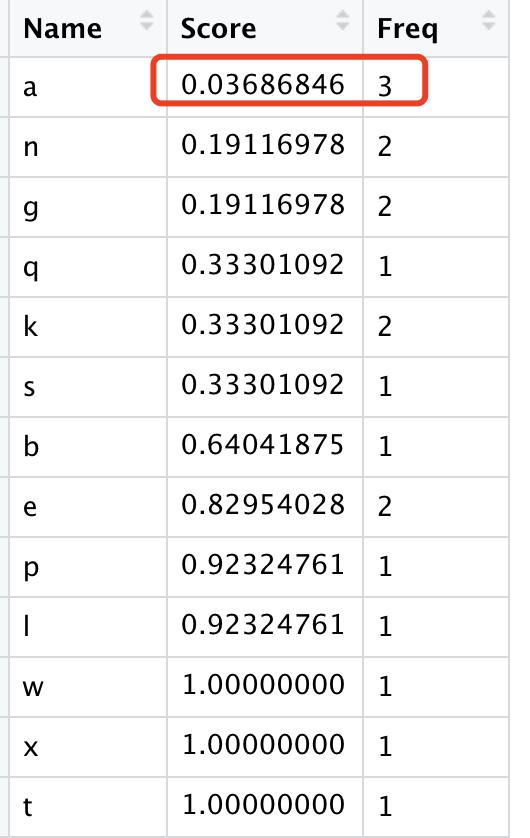
可以看到,a这个字母在3次随机抽样都抽到了,所以它的 exact p-value 非常小,就是统计学非常显著啦!
然后,其余的出现了两次的字母就比较多了,它们的得分之所以有区别,就在于它们的排序。
- n和g都是出现两次,而且排名很靠前,所以p值是0.19,马马虎虎。
- k出现了两次,q出现一次,而且都有一个在各自的抽样场合排名第一,k的另外一次在最后面所以权重很低,所以p值是0.33,很差了。
- 至于e和y,虽然也是出现了两次,但是都排名超级靠后,所以p值也很辣鸡,接近于1了。
总结一下, aggregateRanks函数其实就是对多个排好序的基因集,进行求交集的同时还考虑一下它们的排序情况。总体上来说,就是挑选那些在多个数据集都表现差异的基因,并且每次差异都排名靠前的那些。
真实多个GEO数据集的差异分析结果的aggregateRanks函数
下面是付费代码,有点难度,但是超级实用!
library(clusterProfiler)
set.seed(123456789)
data(geneList, package="DOSE")
head(geneList)
deg=data.frame(gene=names(geneList),
logFC=as.numeric(geneList),
P.Value=0)
head(deg)
# 大家的deg 来源于真实的差异分析
deg2=deg;deg2$gene=sample(deg$gene,length(deg2$gene))
deg3=deg;deg3$gene=sample(deg$gene,length(deg2$gene))
deg4=deg;deg4$gene=sample(deg$gene,length(deg2$gene))
get_up <- function(df){
df$g=ifelse(df$P.Value>0.01,'stable', #if 判断:如果这一基因的P.Value>0.01,则为stable基因
ifelse( df$logFC >2,'up', #接上句else 否则:接下来开始判断那些P.Value<0.01的基因,再if 判断:如果logFC >1.5,则为up(上调)基因
ifelse( df$logFC < -2,'down','stable') )#接上句else 否则:接下来开始判断那些logFC <1.5 的基因,再if 判断:如果logFC <1.5,则为down(下调)基因,否则为stable基因
)
print( table(df$g))
df=df[order(df$logFC,decreasing = T),]
# rownames(df[df$g=='up',])
df[df$g=='up','gene']
}
glist=list(get_up(deg2)
,get_up(deg3)
, get_up(deg4))
ups=aggregateRanks(glist = glist, N = length(unique(unlist(glist))))
tmp=as.data.frame(table(unlist(glist)))
ups$Freq=tmp[match(ups$Name,tmp[,1]),2]
head(ups)
因为是完全随机模拟的3个差异分析结果, 所以得到的aggregateRanks函数整合好的上调基因是没有合格的!
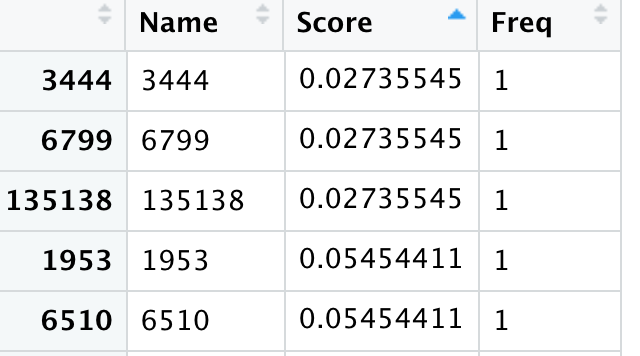
但是假如有合格的基因,就可以进行热图可视化啦,代码也给你:
gs=ups[ups$Score < 0.05,1]
gs
updat=data.frame(deg2=deg2[gs,'logFC'],
deg3=deg3[gs,'logFC'],
deg4=deg4[gs,'logFC'] )
rownames(updat)=gs
library(pheatmap)
head(updat)
pheatmap(updat,display_numbers=T)
当然了,下调基因也是类似的aggregateRanks函数整合,只需要把前面的get_up函数修改一下,可以命名为get_down哦!
你看完差不多就可以学会了,赶紧去处理数据集: GSE7476, GSE13507, GSE37815 and GSE65635 ,验证一下我们的教程吧!
如果这个小技巧,你马上用起来了,也发表了自己的文章,可以参考我们发文章的规范化致谢格式!
We thank Dr.Jianming Zeng(University of Macau), and all the members of his bioinformatics team, biotrainee, for generously sharing their experience and codes.
当然,如果你看完这个细致入微的教程后,居然还不能理解起来,说明你可能是需要手把手教学了,我们生信技能树团队的学习班应该是市面上最适合你的!