最近接到一个粉丝求助,说自己课题组两年前在某公司测的转录组数据,跟着我们的课程学习完了转录组数据分析流程后,终于可以开干了,一条龙流程走完就傻眼了,我们课程授课涉及到的数据集基本上是完美结果,但他们两年前的数据集表现非常的诡异!
fastq测序数据质控的时候
首先fastq测序数据质量控制表格就发现质量差的可怜,而且居然有GC含量的双峰,如下:
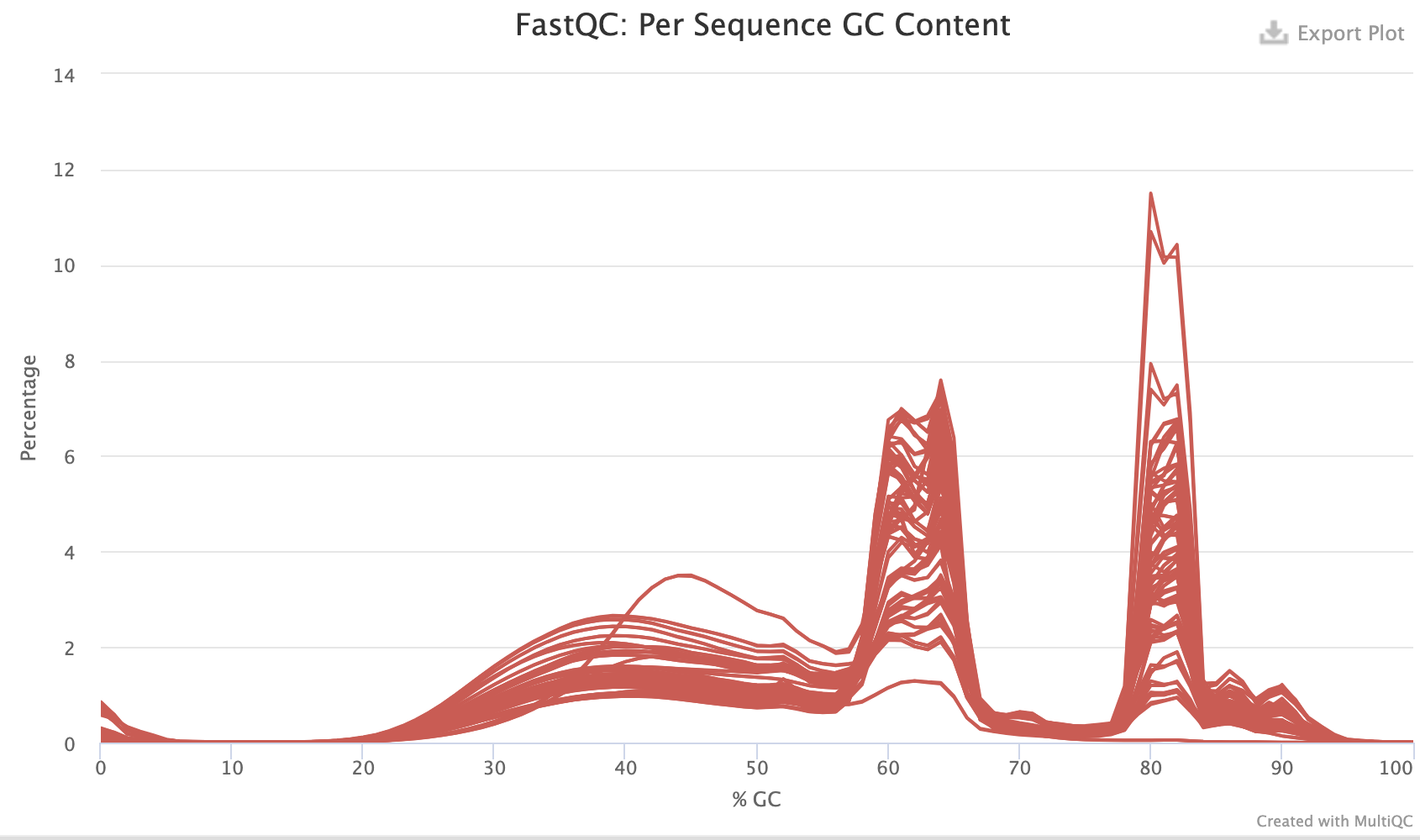
遇到这样的情况,就必须单独看具体的每个样本,上面的GC含量图表是项目里面全部的样本的multiqc汇总图表。
我随机抽一个样本的fastqc报告看了看,如下:
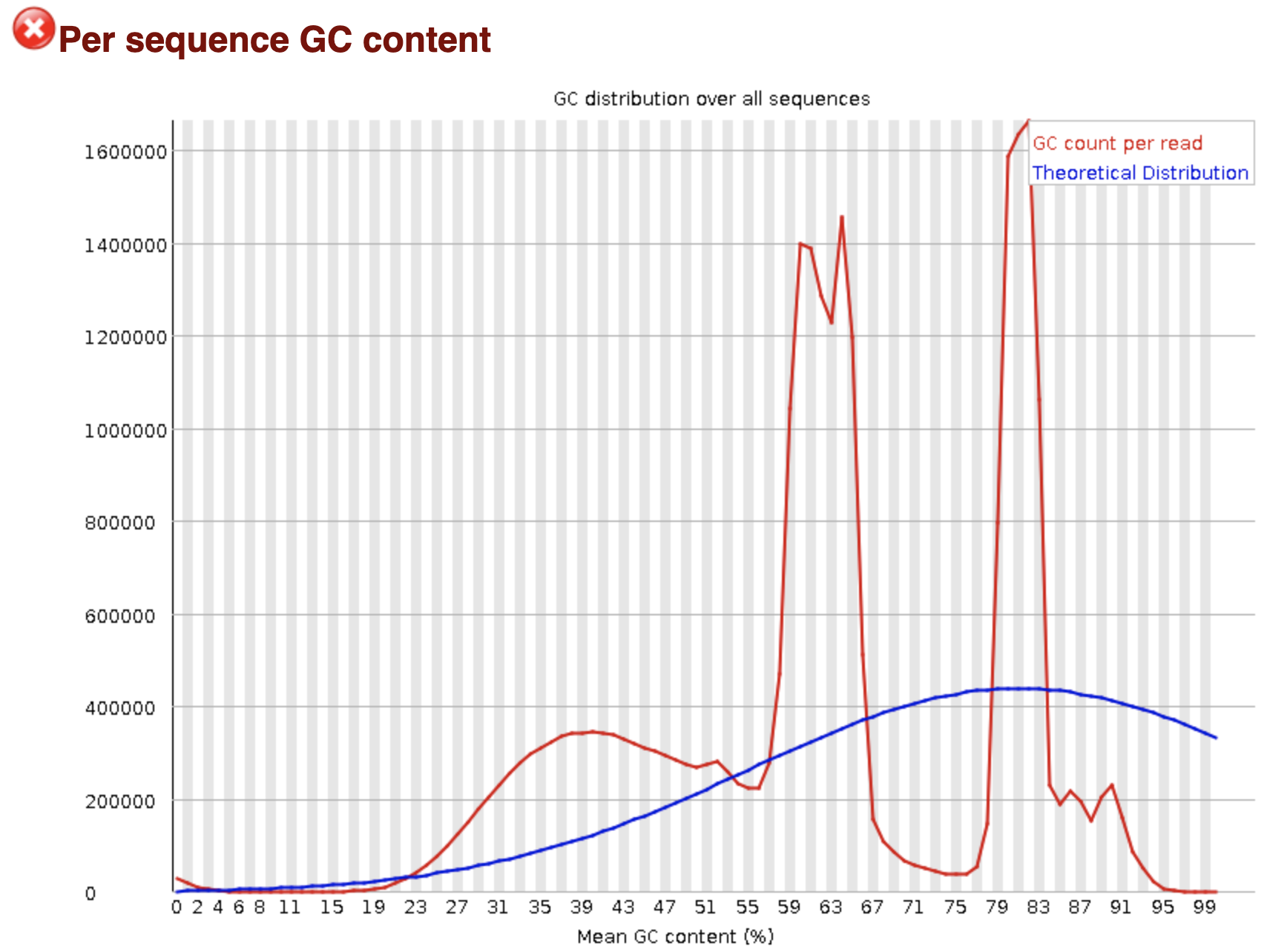
这个59%到67%的GC含量reads有点多啊,就是第一个GC峰值。
然后80%附近GC含量reads也有点多,就是第二个GC含量峰值。
这两个GC含量峰值就是需要解决的问题,正常的RNA-seq数据不会出现这样的情况。
我们有必要仔细看看具体那些动辄一两百万的同一个GC含量的reads是什么,看了看,如下所示:
Sequence Count Percentage Possible Source
CCGGCCCGGAGCGGACGAGCCGCCCCGGCGGTGAACGGGGAGGAGGCGGG 470519 1.6550810978094788 No Hit
CTGGAGTGCAGTGGCTATTCACAGGCGCGATCCCACTACTGATCAGCACG 424455 1.4930479903483649 No Hit
GCGGGGGGCCCGGCGGGGCGTGCGCGTCCGGCGCCGTCCGTCCTTCCGTT 208855 0.7346610077021304 No Hit
CCCGGCCCGGAGCGGACGAGCCGCCCCGGCGGTGAACGGGGAGGAGGCGG 201384 0.7083812806736052 No Hit
GTCCCGGCCCGGAGCGGACGAGCCGCCCCGGCGGTGAACGGGGAGGAGGC 189063 0.6650413641003944 No Hit
GCGCGTGCCTGTAGTCCCAGCTACTCGGGAGGCTGAGGTGGGAGGATCGC 185766 0.6534439527748628 No Hit
GGGGGCCCGGCGGGGCGTGCGCGTCCGGCGCCGTCCGTCCTTCCGTTCGT 184881 0.6503309078785644 No Hit
GGCGCGTGCCTGTAGTCCCAGCTACTCGGGAGGCTGAGGTGGGAGGATCG 184406 0.6486600645726416 No Hit
CGGCGGGGCGTGCGCGTCCGGCGCCGTCCGTCCTTCCGTTCGTCTTCCTC 175251 0.6164567583290131 No Hit
GGCGCGTGCCTGTAGTCCCAGCTACTCGGGAGGCTGAGGCTGGAGGATCG 169817 0.597342310909256 No Hit
GCGCGTGCCTGTAGTCCCAGCTACTCGGGAGGCTGAGGCTGGAGGATCGC 169282 0.5954604137120588 No Hit
CGCGTGCCTGTAGTCCCAGCTACTCGGGAGGCTGAGGTGGGAGGATCGCT 167647 0.5897091951748297 No Hit
CGGGGGGCCCGGCGGGGCGTGCGCGTCCGGCGCCGTCCGTCCTTCCGTTC 161679 0.5687163681227297 No Hit
CCGGCGGGGCGTGCGCGTCCGGCGCCGTCCGTCCTTCCGTTCGTCTTCCT 159369 0.560590793308663 No Hit
GGACGAGCCGCCCCGGCGGTGAACGGGGAGGAGGCGGGAACCGAAGAAGC 155126 0.5456657656307039 No Hit
CGCGTGCCTGTAGTCCCAGCTACTCGGGAGGCTGAGGCTGGAGGATCGCT 152267 0.5356090477114759 No Hit
CCGGAGCGGACGAGCCGCCCCGGCGGTGAACGGGGAGGAGGCGGGAACCG 150767 0.5303327004296142 No Hit
GGCGGGGCGTGCGCGTCCGGCGCCGTCCGTCCTTCCGTTCGTCTTCCTCC 131952 0.4641497176907975 No Hit
GCTATTCACAGGCGCGATCCCACTACTGATCAGCACGGGAGTTTTGACCT 128737 0.45284074668334084 No Hit
CCAGGCTGGAGTGCAGTGGCTATTCACAGGCGCGATCCCACTACTGATCA 124272 0.4371348196076663 No Hit
CGTGCCTGTAGTCCCAGCTACTCGGGAGGCTGAGGTGGGAGGATCGCTTG 123775 0.43538658987494283 No Hit
这个时候,聪明的读者应该是知道,可以去blast这些序列看看到底是啥情况。
比对的时候
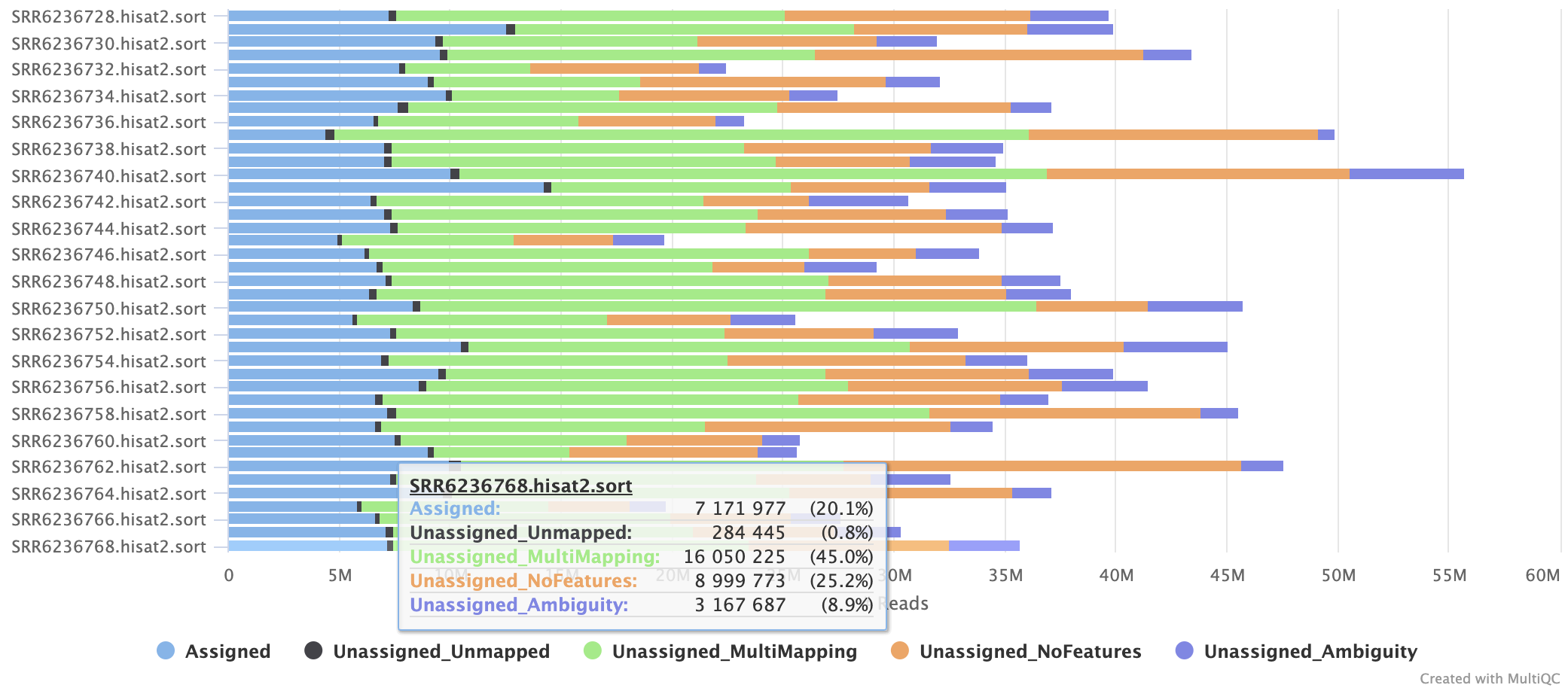
同样的,比对后也使用multiqc汇总,可以看到,多比对情况有点多。一般来说RNA-seq数据,会采取PE100或者PE150的策略,这样的长度其实是很难发生基因组的多比对情况的。
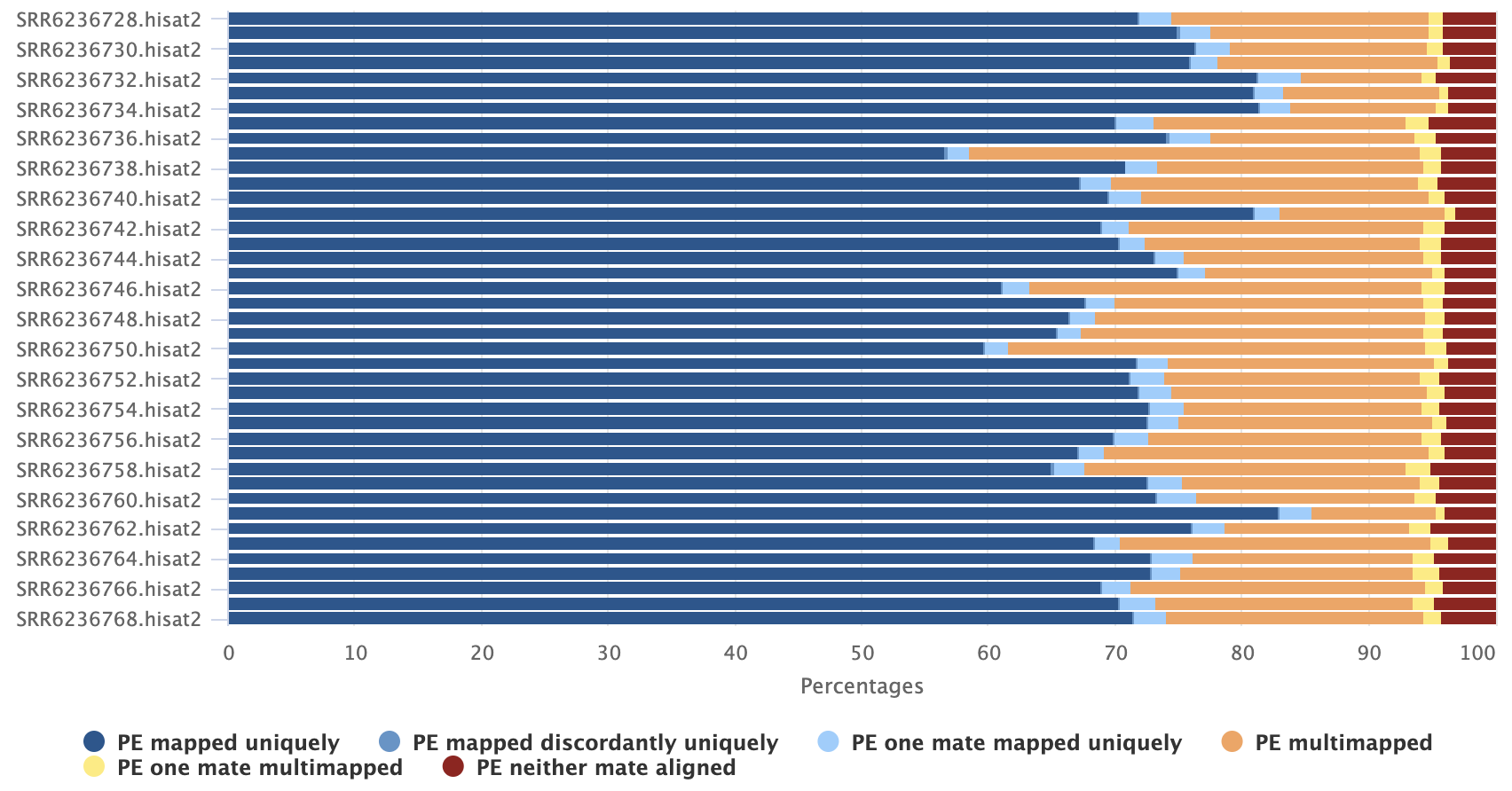
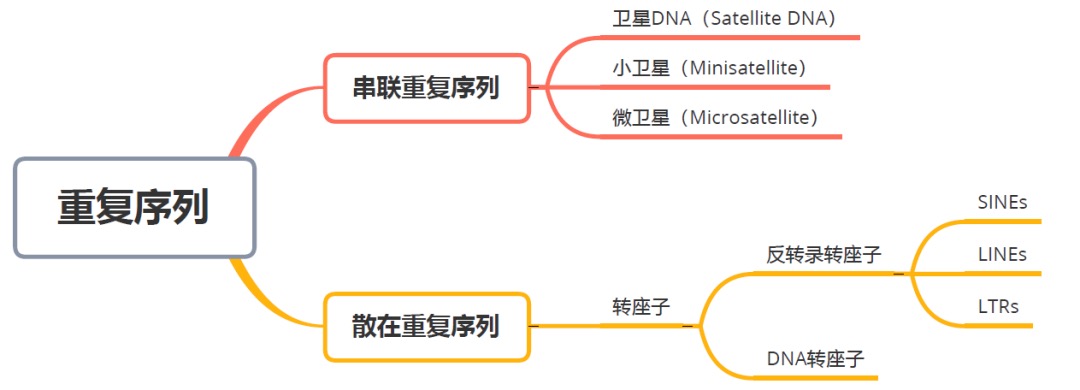
这个时候可以思考一下会不会是基因组的重复序列或集中成簇,根据分布把重复序列分为分散重复序列(Interpersed repeat)和串联重复序列(Tendam repeat)两大类,然后根据两大类中重复序列长度在细分为几个子类。
定量的时候
转录组数据比对后,就需要定量,这个时候的质量控制也很重要。
如果你对转录组的基本分析还有问题,可以看我们的教程合辑:
-
上游分析视频以及代码资料在:https://share.weiyun.com/5QwKGxi
-
下游主要是基于counts矩阵的标准分析的代码 https://share.weiyun.com/50hfuLi
如果你是Linux和R语言基础知识不熟练, 就需要自己恶补啦。计算机基础知识,我把它粗略的分成基于R语言的统计可视化,以及基于Linux的NGS数据处理:
逼自己一把,把R的知识点路线图搞定,如下:
- 了解常量和变量概念
- 加减乘除等运算(计算器)
- 多种数据类型(数值,字符,逻辑,因子)
- 多种数据结构(向量,矩阵,数组,数据框,列表)
- 文件读取和写出
- 简单统计可视化
- 无限量函数学习
Linux的6个阶段也跨越过去 ,一般来说,每个阶段都需要至少一天以上的学习:
- 第1阶段:把linux系统玩得跟Windows或者MacOS那样的桌面操作系统一样顺畅,主要目的就是去可视化,熟悉黑白命令行界面,可以仅仅以键盘交互模式完成常规文件夹及文件管理工作。
- 第2阶段:做到文本文件的表格化处理,类似于以键盘交互模式完成Excel表格的排序、计数、筛选、去冗余,查找,切割,替换,合并,补齐,熟练掌握awk,sed,grep这文本处理的三驾马车。
- 第3阶段:元字符,通配符及shell中的各种扩展,从此linux操作不在神秘!
- 第4阶段:高级目录管理:软硬链接,绝对路径和相对路径,环境变量
- 第5阶段:任务提交及批处理,脚本编写解放你的双手
- 第6阶段:软件安装及conda管理,让linux系统实用性放飞自我