前面我带领大家通过IMGT数据库认知免疫组库,而且也一起从IMGT数据库下载免疫组库相关fasta序列,免疫组库重要的研究对象就是分成BCR的IGH,IGK,IGL这3类,以及TCR的TRA,TRB,TRD,TRG,它们各自都有V,D(可选),J,C基因。
接下来又认识了免疫组库测序数据,知道了免疫组库测序数据的一些特性,现在就面临免疫组库数据分析流程的搭建啦,这个其实非常复杂, 今天我只能勉强介绍一下使用igblast进行免疫组库分析,希望大家能跟上来。其实igblast这个软件早在六年前我就介绍过,差不多是重新看着自己的教程,一点一滴复现了一遍。真的要吹爆生信技能树和生信菜鸟团教程,极大的便利了生信工程师的工作。
igblast因为是ncbi出品,所以在免疫组库分析领域还算是使用频率较高的,值得注意的是igblast软件虽然下载即可使用,但是软件用法超级复杂,软件输出的结果文件需要耗费至少五六个小时去理解。
首先看看网页版igblast
官网是:https://www.ncbi.nlm.nih.gov/igblast/
简单的复制粘贴一条免疫组库测序数据以FASTA格式粘贴到网页的输入框即可,默认的物种是人类:
TGTGCCAGCAGCTTGGCCCGAGAAGGGATTGAAAACACCATATATTTT
我们这里仍然是使用在前面我们认识的免疫组库测序数据,是人类的,MiSeq测序仪,PE300测序策略,TRB,DNA测序,进行示范。
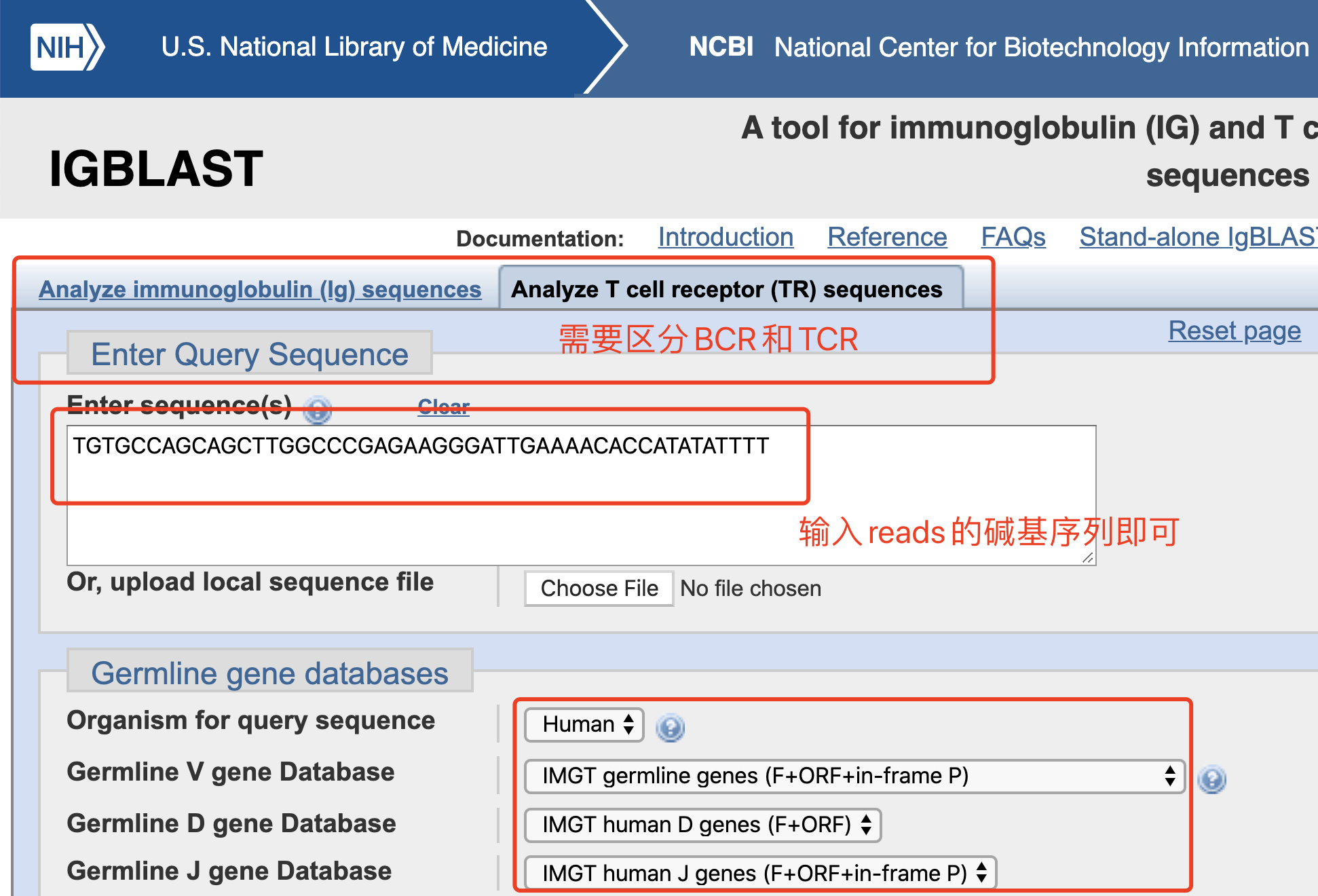
因为是TRB,所以选择
Database: imgt.TR.Homo_sapiens.V.f.orf.p; imgt.TR.Homo_sapiens.D.f.orf;
imgt.TR.Homo_sapiens.J.f.orf.p
376 sequences; 82,149 total letters
输出结果如下:
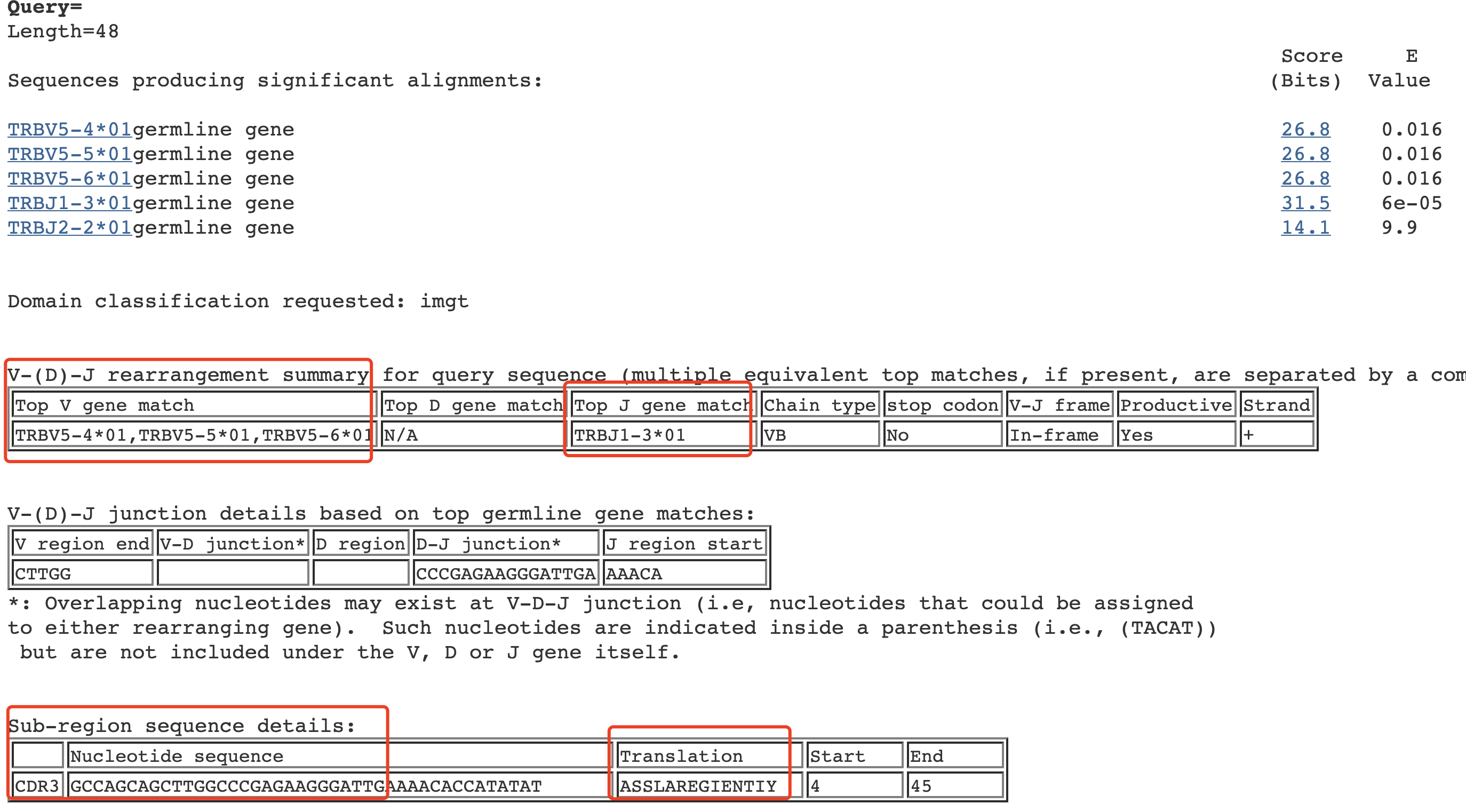
因为我们输入的碱基序列只有48个碱基,所以比对结果里面,对V基因来说,TRBV5-401,TRBV5-501的得分是一样的。然后最重要的是CDR3序列,包括核苷酸序列和氨基酸序列。
软件安装及数据库文件准备
首先是软件,因为是二进制,所以下载解压即可使用
在 ftp://ftp.ncbi.nih.gov/blast/executables/igblast/release/ 查看最新版软件下载
mkdir -p ~/biosoft/igblast
cd ~/biosoft/igblast
# 软件是39M,下载速度可能会比较慢
wget -c ftp://ftp.ncbi.nih.gov/blast/executables/igblast/release/1.9.0/ncbi-igblast-1.9.0-x64-linux.tar.gz
tar -xzf ncbi-igblast-1.9.0-x64-linux.tar.gz
# 因为我电脑是Mac,所以这个 ncbi-igblast-1.9.0-x64-linux.tar.gz 没有用。
# 然后因为网速问题,我其实最后选择了conda安装igblast-1.7.0
conda install igblast
数据库文件也是 ftp://ftp.ncbi.nih.gov/blast/executables/igblast/release/

这3 个文件夹都需要下载.
使用igblast进行序列比对
我们这里仍然是使用在前面我们认识的免疫组库测序数据,是人类的,MiSeq测序仪,PE300测序策略,TRB,DNA测序,进行示范。
拼接双端测序数据
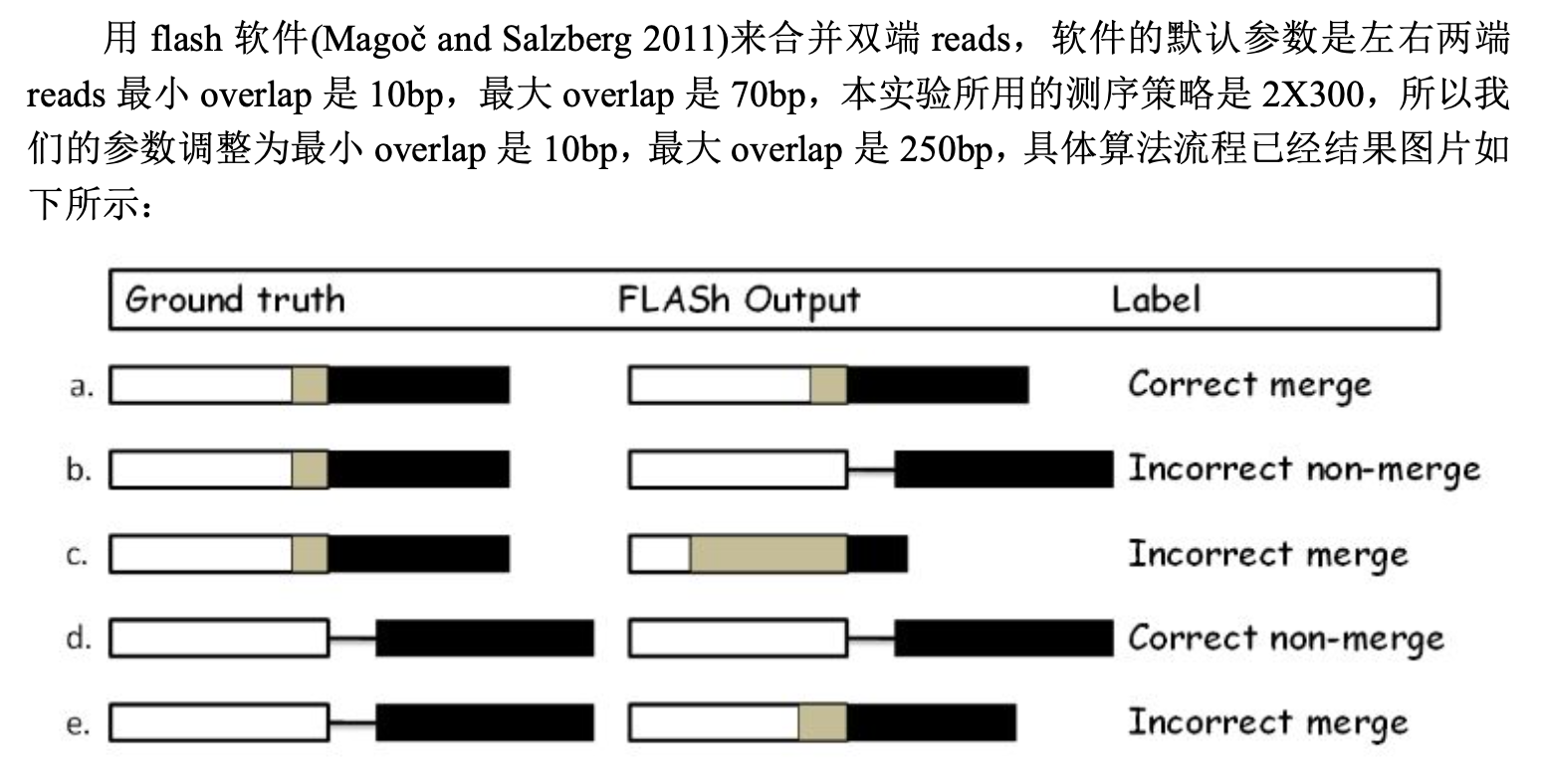
我们提到过,对于PE序列,首先需要拼接,这里我们采用我五年前的流程,就是FLASH软件。
FLASH (F ast L ength A djustment of SH ort reads) is a very fast and accurate software tool to merge paired-end reads from next-generation sequencing experiments.
FLASH软件的全称为Fast Length Adjustment of Short Reads,自2011年被发表在《Bioinformatics》期刊上以来,该软件被引用量超千次,其能够借助PE clean reads之间的overlap,将测序产生的paired-end reads快速拼接为DNA片段。如果两条reads的长度总和大于原始测序片段的总长度就可以使用FLASH进行拼接,但是不能拼接不存在overlap的paired-end reads。
FLASH软件可以对PE测序,左右两端reads进行序列拼接,示意图如下:

mkdir -p ~/biosoft/FLASH
cd ~/biosoft/FLASH
wget http://ccb.jhu.edu/software/FLASH/FLASH-1.2.11-Linux-x86_64.tar.gz
tar zxvf FLASH-1.2.11-Linux-x86_64.tar.gz
./FLASH-1.2.11-Linux-x86_64/flash
# Usage: flash [OPTIONS] MATES_1.FASTQ MATES_2.FASTQ
# Run `./FLASH-1.2.11-Linux-x86_64/flash --help | less' for more information.
本来我以为这个软件在Linux可以使用,那么在Mac就应该是类似,但实际情况下被打脸了。所以我还是conda安装吧,至少在Mac电脑得这样。
conda install -c bioconda flash
然后把前面我们认识的免疫组库测序数据进行左右fastq文件的合并!
命令如下:
cd ~/data/public/TRB/test/
flash raw/ERR3445170_1.fastq.gz raw/ERR3445170_2.fastq.gz -M 250
大部分参数都使用默认值即可,其中 -M 指的是拼接时overlap区的最长bp阈值,默认65,我们这里调整为250;
FLASH拼接默认输出5个结果文件:
- output.extendeFrags.fastq 为拼接后的扩增片段序列文件;
- output.flash.log 为日志文件,详细记录了拼接过程中的参数和拼接统计的数据;
- output.hist 为拼接后的reads长度的统计信息文件;
- output.histogram 为拼接后的reads长度直方图文件;
- output.notCombined_1.fastq 为拼接不上的reads1序列文件;
- output.notCombined_2.fastq 为拼接不上的reads2序列文件;
其中软件运行的log日志,需要注意的是:
[FLASH] Read combination statistics:
[FLASH] Total pairs: 84833
[FLASH] Combined pairs: 30753
[FLASH] Uncombined pairs: 54080
[FLASH] Percent combined: 36.25%
这个比例并不高,调整flash软件参数可以改进
flash raw/ERR3445170_1.fastq.gz raw/ERR3445170_2.fastq.gz -M 250
重要参数解释如下:
- -m 拼接时overlap区的最小bp阈值,默认10bp;
- -M 拼接时overlap区的最长bp阈值,默认65,我们这里调整为250;
- -x overlap区允许的最大碱基错配比率(最大碱基错配数目/overlap区长度),默认为0.25;
- -p 碱基质量值类型,phred64或者33,默认是33;
- -r reads长度;
- -f 片段长度,也就是测序的文库大小;
- -s 文库的偏差;
- -o 输出文件前缀;
- -t 设置线程数,默认为1,FLASH软件支持多线程,速度快;
或者应该是先去除接头和低质量reads,比如走trim_galore 流程。
conda create -n qc -y
source activate qc
conda install -y trim-galore
trim_galore -q 25 --phred33 --length 36 --stringency 3 --paired -o clean raw/ERR3445170_1.fastq.gz raw/ERR3445170_2.fastq.gz
# 然后再使用flash对PE数据进行拼接
flash raw/ERR3445170_1.fastq.gz raw/ERR3445170_2.fastq.gz -M 250 -o clean
这次的log日志如下:
[FLASH] Read combination statistics:
[FLASH] Total pairs: 84047
[FLASH] Combined pairs: 81600
[FLASH] Uncombined pairs: 2447
[FLASH] Percent combined: 97.09%
可以看到,这个拼接比率增加的非常棒!
构建人类的免疫组库数据库
首先需要研读从IMGT数据库下载免疫组库相关fasta序列,我们这里举例的是TRB测序,所以下载TRB的V,D,J的FASTA文件。
mkdir -p ~/biosoft/igblast/imgt
cd ~/biosoft/igblast/imgt
wget http://www.imgt.org/download/V-QUEST/IMGT_V-QUEST_reference_directory/Homo_sapiens/TR/TR{A,B,D,G}{V,J}.fasta
wget http://www.imgt.org/download/V-QUEST/IMGT_V-QUEST_reference_directory/Homo_sapiens/TR/TR{B,D}D.fasta
然后对下载TRB的V,D,J的FASTA文件进行igblast索引构建。
cd ~/biosoft/igblast
perl edit_imgt_file.pl imgt/TRBV.fasta > database/human_TRBV.fa
makeblastdb -parse_seqids -dbtype nucl -in database/human_TRBV.fa
perl edit_imgt_file.pl imgt/TRBD.fasta > database/human_TRBD.fa
makeblastdb -parse_seqids -dbtype nucl -in database/human_TRBD.fa
perl edit_imgt_file.pl imgt/TRBJ.fasta > database/human_TRBJ.fa
makeblastdb -parse_seqids -dbtype nucl -in database/human_TRBJ.fa
其它物种类似的处理方式。
运行igblast
接下来才是真正的igblast程序运行,有了fasta序列和免疫组库的TRB的V,D,J参考序列。
conda install -c bioconda seqkit
seqkit fq2fa clean.extendedFrags.fastq > in.fa
# 因为igblastn的输入数据需要是fasta格式
igblastn \
-germline_db_V ~/biosoft/igblast/database/human_TRBV.fa \
-germline_db_D ~/biosoft/igblast/database/human_TRBD.fa \
-germline_db_J ~/biosoft/igblast/database/human_TRBJ.fa \
-domain_system imgt \
-organism human \
-ig_seqtype TCR \
-auxiliary_data ~/biosoft/igblast/optional_file/human_gl.aux \
-show_translation \
-outfmt 7 \
-num_threads 4 \
-query in.fa \
-out test.blast.output.7
得到的结果如下:
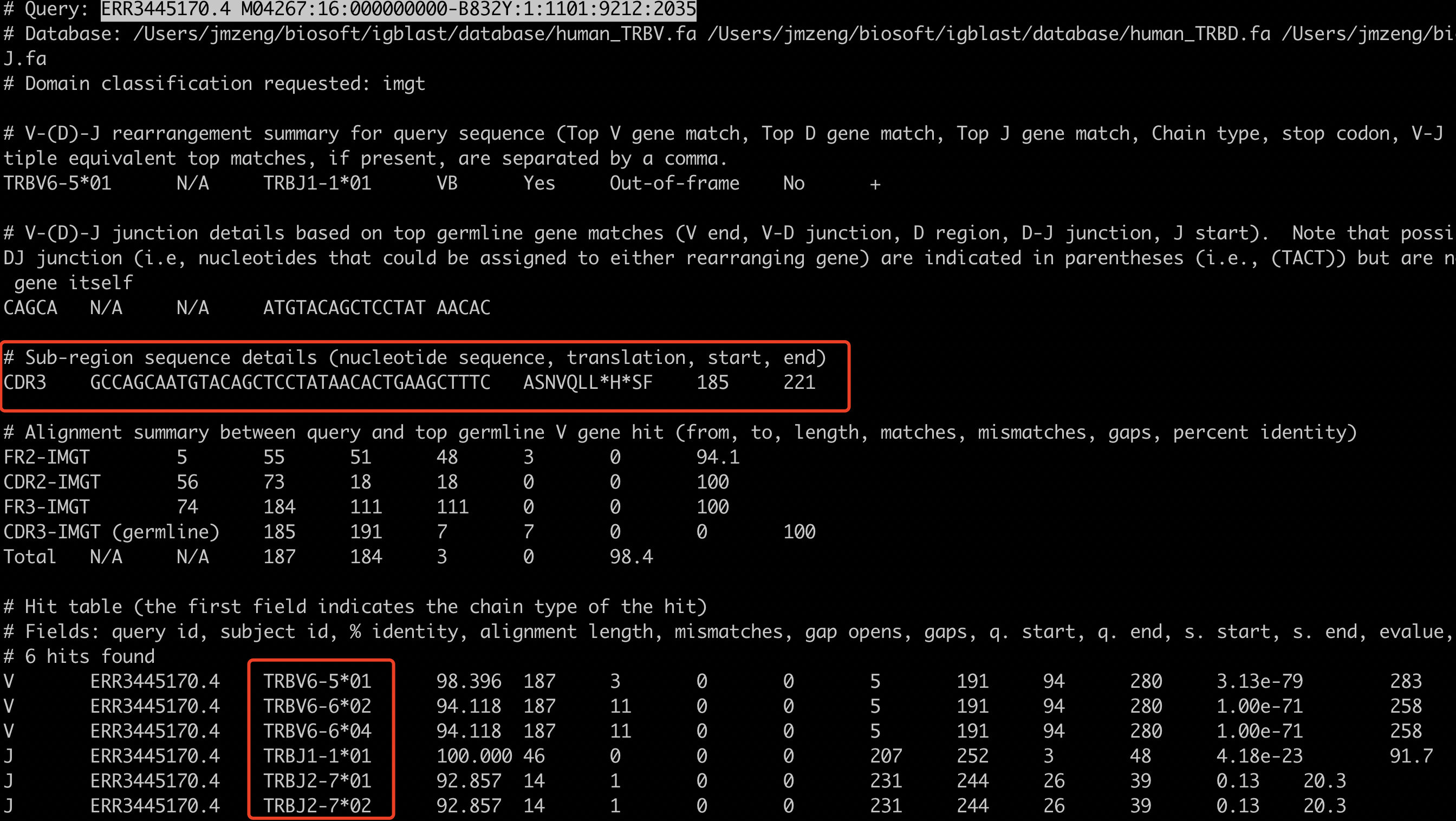
免疫组库的测试数据
有一个工具,是Change-O
- Change-O is a collection of tools for processing the output of V(D)J alignment tools, assigning clonal clusters to immunoglobulin (Ig) sequences, and reconstructing germline sequences.
提供了测试数据:
- We have hosted a small example data set resulting from the Roche 454 example workflow described in the pRESTO documentation. In addition to the example FASTA files, we have included the standalone IgBLAST results.
大家可以试试看,使用我们上面讲解的igblastn命令,分析这个数据集。
参考: