前面我在单细胞天地的教程:10X单细胞转录组理论上有3个文件才能被读入R进行seurat分析,预告了一个粉丝遇到的疑难点,数据集GSE127465里面明明是可以下载到看起来是10X标准的3个文件,但是的确没办法读入到R里面进行seurat流程。
我就帮他仔细看了看,发现 GSE127465_human_counts_normalized_54773x41861.mtx.gz 没有啥问题,妥妥的稀疏矩阵,但是 GSE127465_human_cell_metadata_54773x25.tsv.gz 里面很明显并不是普通的细胞信息,里面五花八门。我也是非常纳闷,当时见面会的场面一度十分尴尬,这背后有一个小故事:
本来吧,我是飞去西安参加婚礼,有一个单细胞课程学员联系过去西安请吃饭。就随便去他们实验室拜访一下,结果去了才知道是大场面,整个实验室出动了来跟我交流,而且还现场问了这个数据集的下游分析问题。关键是我参加婚礼没带电脑啊,只能是使用学生的“辣鸡”电脑,现场手写代码一个小时。
我想了想,只能是读入这个稀疏矩阵了,放弃另外的两个文件,细胞信息和基因信息,可以后期加上。代码如下:
rm(list=ls())
options(stringsAsFactors = F)
library(Seurat)
library(Matrix)
x=readMM("raw/GSE127465_human_counts_normalized_54773x41861.mtx")
x
colnames(x)
rownames(x)
dim(x)
这个稀疏矩阵文件gz格式压缩是500M,解压后1.4G,我本来是想把它转为普通矩阵然后构建seurat对象,但是考虑到其是54773x41861的信息,我就放弃了 ,毕竟是在学生的“辣鸡”电脑,才8G电脑。
# 这个 x 是前面 readMM函数读入的稀疏矩阵
# 如果下面的代码保存,就说明readMM读入的稀疏矩阵,需要首先转为普通矩阵
CreateSeuratObject(counts = x,
meta.data =meta,
min.cells = 3,
min.features = 200,
project = "GSE127465")
然后就可以进行后续分析了,走seurat流程。
其实我可以自己构造另外两个文件,然后把3个文件存放在同一个文件夹,然后使用下面的代码进行读取
rm(list=ls())
options(stringsAsFactors = F)
library(Seurat)
sce1 <- CreateSeuratObject(Read10X('../10x-results/WT/'),
"GSE127465")
我回去后一直在思考,为什么这个作者并没有提供标准的3个文件,而仅仅是一个稀疏矩阵呢?
后来我看了看文献,我勒个去,这个根本就不是10x的单细胞转录组,而是inDrop技术。
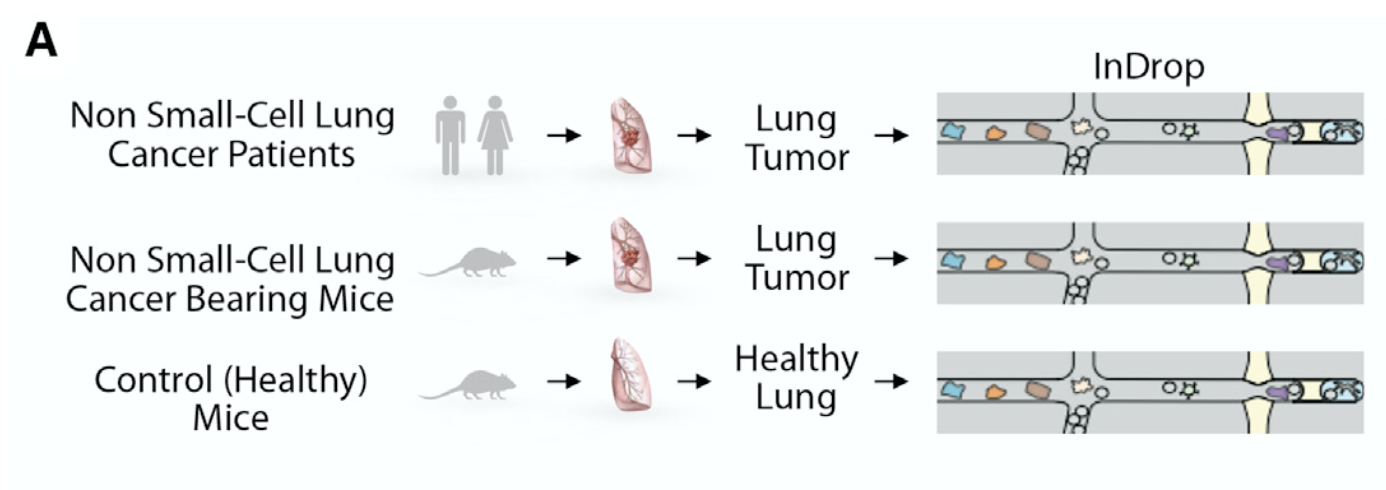
所以全文压根就没有10X的事情,仅仅是Matrix包的readMM函数即可。在数据集GSE127465配套的文章里面也写的很清楚:

关于那个indrop.py的流程,我们在单细胞天地也分享过。