前面我已经把全网第一个单细胞课程(基础课程)免费公布在了B站:https://www.bilibili.com/video/av38741055
- 结业考核20题:https://mp.weixin.qq.com/s/lpoHhZqi-_ASUaIfpnX96w
- 课程配套资料文档在:https://docs.qq.com/doc/DT2NwV0Fab3JBRUx0
虽然是免费的, 但是学员关于课程的提问,我还是会尽我所能帮忙的。当然,受限于时间和精力,只能是挑选重点和普适性的有价值的问题,比如其中一个问题是:
我发现我的目的基因主要分布在B细胞群,想问一下能不能通过是否表达目的基因将B细胞分成两群,再寻找差异表达基因呀?我看seurat包中,findmarkers的函数只要能找不同cluster 间的差异基因?
这个问题有两个解决方案,第一个把已经划分为B细胞群的那些细胞的表达矩阵,重新走seurat流程,看看这个时候它们是否根据有没有表达目的基因来进行分群,如果有,就可以使用 findmarkers函数。
另外一个解决方案,就是人为划分,然后强行走seurat标准流程。
首先看看 seurat标准流程
首先看看我们的seurat标准流程,自己去GEO数据库下载GSE125616的GSE125616_READ_COUNT_table_lvbo_TE_gencode.txt.gz文件,然后走下面的代码即可:
rm(list=ls())
options(stringsAsFactors = F)
library(data.table)
a=fread('GSE125616_READ_COUNT_table_lvbo_TE_gencode.txt.gz',
header = T,sep = '\t',data.table = F)
hg=a$Geneid
dat=a[,7:ncol(a)]
rownames(dat)=hg
hg[grepl('^MT-',hg)]
head(hg)
library(stringr)
colnames(dat)
colnames(dat)=str_split(colnames(dat),'_',simplify = T)[,1]
meta=as.data.frame(str_split(colnames(dat),'_',simplify = T)[,1])
colnames(meta)=c('cell name')
rownames(meta)=colnames(dat)
library(Seurat)
pbmc <- CreateSeuratObject(counts = dat,
meta.data = meta ,
min.cells = 5, min.features = 1000,
project = "test")
pbmc
pbmc[["percent.mt"]] <- PercentageFeatureSet(pbmc, pattern = "^MT-")
sce=pbmc
sce <- NormalizeData(sce, normalization.method = "LogNormalize", scale.factor = 10000)
GetAssay(sce,assay = "RNA")
sce <- FindVariableFeatures(sce, selection.method = "vst", nfeatures = 2000)
# 步骤 ScaleData 的耗时取决于电脑系统配置(保守估计大于一分钟)
sce <- ScaleData(sce)
sce <- RunPCA(object = sce, pc.genes = VariableFeatures(sce))
sce <- FindNeighbors(sce, dims = 1:15)
sce <- FindClusters(sce, resolution = 0.2)
table(sce@meta.data$RNA_snn_res.0.2)
set.seed(123)
sce <- RunTSNE(object = sce, dims = 1:15, do.fast = TRUE)
DimPlot(sce,reduction = "tsne",label=T)
phe=data.frame(cell=rownames(sce@meta.data),
cluster =sce@meta.data$seurat_clusters)
head(phe)
table(phe$cluster)
可以看到, 走默认代码,划分的是5个亚群:
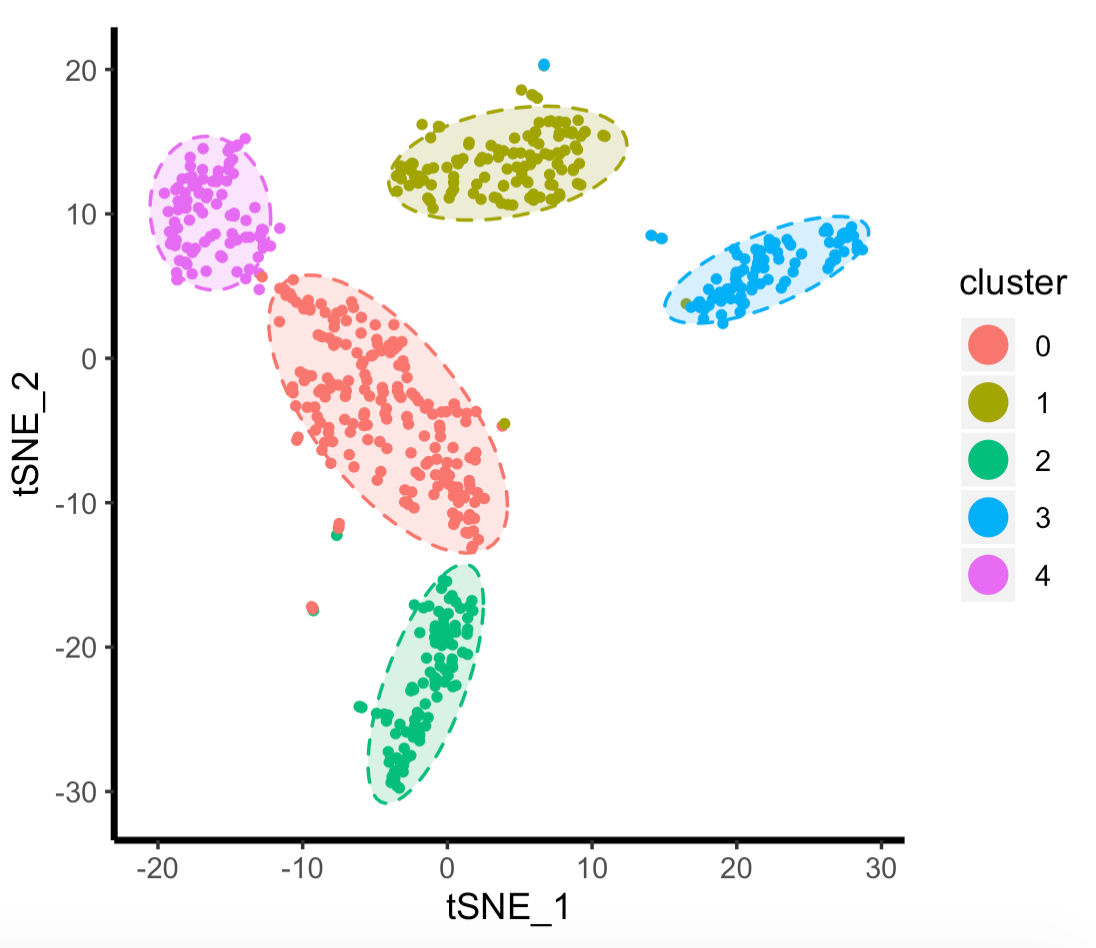
这个时候,学员的问题可以简化为,不满意上图里面的0那个亚群,觉得它可以继续划分,比如根据感兴趣的目的基因,强行把0那个红色亚群继续划分为高低表达(或者是否表达)目的基因的亚群。
先熟悉FindMarkers函数
通常,我们使用FindMarkers函数针对感兴趣的细胞亚群,去寻找它与其它所有的亚群,表达有差异的基因,代码如下:
markers_df <- FindMarkers(object = sce, ident.1 = 0, min.pct = 0.25)
print(x = head(markers_df))
markers_genes = rownames(head(x = markers_df, n = 5))
VlnPlot(object = sce, features =markers_genes,log =T )
FeaturePlot(object = sce, features=markers_genes )
markers_df <- FindMarkers(object = sce, ident.1 = 1, min.pct = 0.25)
print(x = head(markers_df))
markers_genes = rownames(head(x = markers_df, n = 5))
VlnPlot(object = sce, features =markers_genes,log =T )
FeaturePlot(object = sce, features=markers_genes )
如果你仔细看这个FindMarkers函数的说明书,它其实是可以给定两个亚群,然后单独比较这两个亚群的。
## Default S3 method:
FindMarkers(
object,
slot = "data",
counts = numeric(),
cells.1 = NULL,
cells.2 = NULL,
features = NULL,
reduction = NULL,
logfc.threshold = 0.25,
test.use = "wilcox",
min.pct = 0.1,
min.diff.pct = -Inf,
verbose = TRUE,
only.pos = FALSE,
max.cells.per.ident = Inf,
random.seed = 1,
latent.vars = NULL,
min.cells.feature = 3,
min.cells.group = 3,
pseudocount.use = 1,
...
)
所以,我们的目标就是把两个亚群信息,给到这个FindMarkers函数即可。当然了,这个差异分析结果表格也是需要理解的:
avg_logFC: log fold-chage of the average expression between the two groups. Positive values indicate that the gene is more highly expressed in the first group
pct.1: The percentage of cells where the gene is detected in the first group
pct.2: The percentage of cells where the gene is detected in the second group
p_val_adj: Adjusted p-value, based on bonferroni correction using all genes in the dataset
主要是借助统计可视化来。
根据高低表达(或者是否表达)目的基因划分亚群
其实在这个FindMarkers函数的说明书里面,就有一个现成的例子:
# Take all cells in cluster 2, and find markers that separate cells in the 'g1' group (metadata
# variable 'group')
markers <- FindMarkers(pbmc_small, ident.1 = "g1", group.by = 'groups', subset.ident = "2")
head(x = markers)
所以我们也是需要给单细胞数据对象,制作一个新的分组即可;
# 下面的 TP53 > 10 仅仅是我举例方便,你有自己感兴趣的基因,自己感兴趣的基因表达量阈值
highCells=colnames(subset(x = sce, subset = TP53 > 10, slot = 'counts'))
highORlow=ifelse(colnames(sce) %in% highCells,'high','low')
table(highORlow)
sce@meta.data$highORlow=highORlow
markers <- FindMarkers(sce, ident.1 = "high",
group.by = 'highORlow',
subset.ident = "0")
head(x = markers)
是不是很简单啊?
为什么我会挑选这个问题来解答呢,其实就是督促大家读说明书。