五年前我在生信菜鸟团博客分享了 一篇文章学会miRNA-seq分析 ,使用 RNA expression profiling of human iPSC-derived cardiomyocytes in a cardiac hypertrophy model. PLoS One 2014;9(9):e108051. PMID: 25255322 文章里面的 https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE60292 数据集,2个分组,共6个样本。
那个时候举例使用的是bowtie2软件比对miRNA的reads到miRBase里面的miRNA序列文件,以及hg38参考基因组,两个策略。后来也看了看很多公司报告,发现大多集中于下游分析,就是拿到了miRNA表达矩阵后的,包括差异分析,靶基因等等。如下所示:
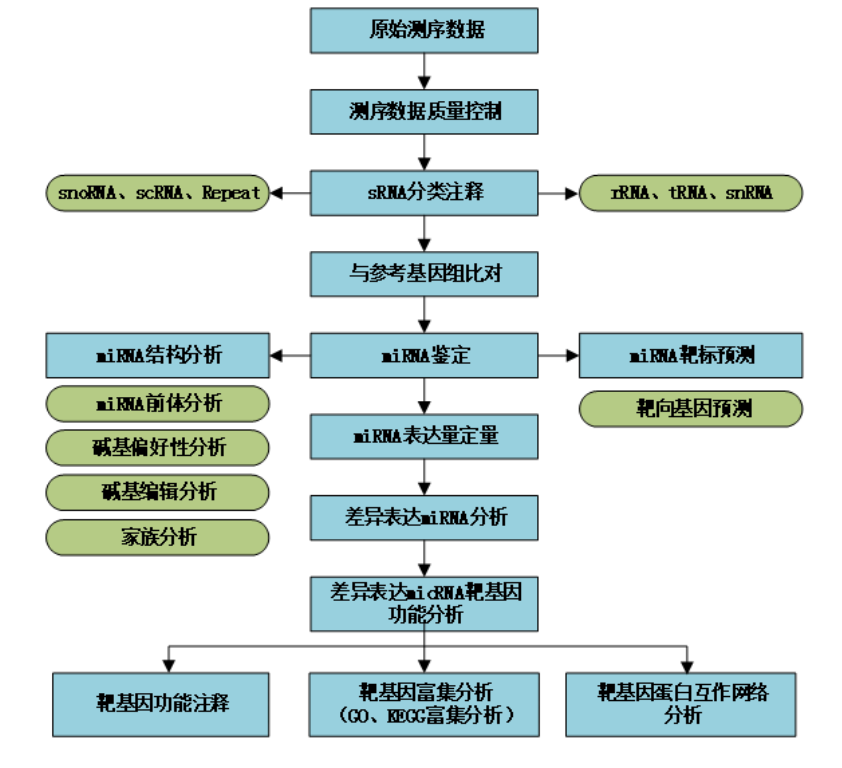
我最近在生信技能树分享了几个miRNA的靶向基因的查询工具,分别是:
- microRNAs靶基因数据库哪家强
- 使用miRNAtap数据源提取miRNA的预测靶基因结果
- 对miRNA进行go和kegg等功能数据库数据库注释
但是在回看自己五年前的 一篇文章学会miRNA-seq分析 ,发现反而是上游分析并不具备固定的流程,如果上游分析都有疑问,意味着拿到的miRNA表达矩阵本来是有问题的,后续的下游分析也就无从谈起了。
比如发表在Genome Biol. 2014的文章Evidence for the biogenesis of more than 1,000 novel human microRNAs的流程就值得介绍:测序数据质控环节:
- Sequencing was performed on a HiSeq2000 instrument running TruSeq version 3 chemistry for 50 cycles.
- Base calling and quality score calculation was performed from raw intensities using Illumina’s pipeline version 1.8.1.
- The called reads were trimmed with the command line: fastx_trimmer –f 1 –l 36 and low-quality reads discarded with fastx_artifacts_filter using the options –q 10.
- Adapters were clipped using the AdRec.jar program from the seqBuster suite with the following options: java -jar AdRec.jar 1 8 0.3.
如果要发现新的miRNA,需要比对到参考基因组。
- 使用bowtie –f –v 0 –a –m 5 —strata —best; 比对miRNA的FASTA文件到人类参考基因组
- 删除属于annotations of tRNA or rRNA (RepeatMasker hg19)和 known miRNA hairpins (miRBase version 19)的已知miRNA
值得注意的是,small interfering RNA (siRNA), Piwi interacting RNA (piRNA) and microRNAs (miRNAs) 需要区分开来哦,我们现在说的是miRNAs相关的测序数据分析。
但是绝大部分人在处理miRNA测序数据的时候,并不会有那个时间来仔细琢磨这个数据处理流程。所以,如果你仔细看流程,会发现千奇百怪的数据处理。有tophat2比对
在文章 Distinct methylation levels of mature microRNAs in gastrointestinal cancers 可以看到:

但是现在的你,可不能照抄哦,五年前我在生信菜鸟团博客写过一个《RNA-seq流程需要进化啦》,上面分享过:Tophat 首次被发表已经是6年前
Cufflinks也是五年前的事情了
Star的比对速度是tophat的50倍,hisat更是star的1.2倍。
stringTie的组装速度是cufflinks的25倍,但是内存消耗却不到其一半。
Ballgown在差异分析方面比cuffdiff更高的特异性及准确性,且时间消耗不到cuffdiff的千分之一
Bowtie2+eXpress做质量控制优于tophat2+cufflinks和bowtie2+RSEM
Sailfish更是跳过了比对的步骤,直接进行kmer计数来做QC,特异性及准确性都还行,但是速度提高了25倍
kallisto同样不需要比对,速度比sailfish还要提高5倍!!!
bowtie比对第1篇文章
好奇怪,一直有人坚守bowtie,而不是bowtie2,我猜测是不是因为这个bowtie有一个特殊的功能,是bowtie2所不具备的。
A Panel of MicroRNAs as Diagnostic Biomarkers for the Identification of Prostate Cancer
描述如下:

bowtie比对第2篇文章
Expanding the repertoire of miRNAs and miRNA-offset RNAs expressed in multiple myeloma by small RNA deep sequencing

bowtie比对第3篇文章
hsa-miR-9-3p and hsa-miR-9-5p as Post-Transcriptional Modulators of DNA Topoisomerase IIa in Human Leukemia K562 Cells with Acquired Resistance to Etoposide

使用BWA软件的
见发表在 Nucleic Acids Res. 2016 Jan 8的文章 Large-scale profiling of microRNAs for The Cancer Genome Atlas:
- Our adapter-trimming algorithm identified as long an adapter sequence as possible, allowing a number of mismatches that depended on the adapter length found.
- Because the shortest mature miRNA in miRBase v16 is 15 bp, we discarded any trimmed read that was shorter than 15 bp.
- We used BWA-MEM with parameters samse -n 10 to align the remaining reads to a reference genome, which, for most TCGA cancers, was GRCh37
总结一下,目前是bowtie软件来比对miRNA的reads居多。如果大家有趁手的miRNA上游分析流程
欢迎共享哦,比如大家可以看到的tcga数据库的mRNA Analysis Pipeline ,详细代码:
- https://docs.gdc.cancer.gov/Data/Bioinformatics_Pipelines/Expression_mRNA_Pipeline/
太复杂的流程就算了,比如上面提到的发表在 Nucleic Acids Res. 2016 Jan 8; 的文章的流程: - https://github.com/bcgsc/mirna
普通人一辈子也就是处理两三次miRNA数据,并不是TCGA计划那样专业的团队,所以我们仅仅是关心测序reads的清洗问题,接头去除,以及比对的策略。定量之后的表达矩阵分析,反而是很简单的。
欢迎分享,发邮件给我,到 jmzeng1314@163.com文末友情宣传
强烈建议你推荐我们生信技能树给身边的博士后以及年轻生物学PI,帮助他们多一点数据认知,让科研更上一个台阶:
- 生信爆款入门-全球听(买一得五)(第4期),你的生物信息学入门课
- 数据挖掘第2期(两天变三周,实力加量),医学生/临床医师首选技能提高课
- 生信技能树的2019年终总结 ,你的生物信息学成长宝藏
- 2020学习主旋律,B站74小时免费教学视频为你领路,还等什么,看啊!!!