最近有两年前的学生过来寻求合作,让我想想给我tcga数据库过万病人的原始测序数据,我可以做什么方法学的创新。我想把这个问题抛给粉丝:

假设给你tcga数据库过万病人的原始测序数据你可以做什么???
大家应该是都知道,TCGA数据库是目前最综合最全面的癌症病人相关组学数据库,包括:
- DNA Sequencing
- miRNA Sequencing
- Protein Expression array
- mRNA Sequencing
- Total RNA Sequencing
- Array-based Expression
- DNA Methylation
- Copy Number array
知名的肿瘤研究机构都有着自己的TCGA数据库探索工具,比如: - Broad Institute FireBrowse portal, The Broad Institute
- cBioPortal for Cancer Genomics, Memorial Sloan-Kettering Cancer Center
但是现在通常是只能下载到分析过的数据 - http://gdac.broadinstitute.org/runs/stddata__latest/
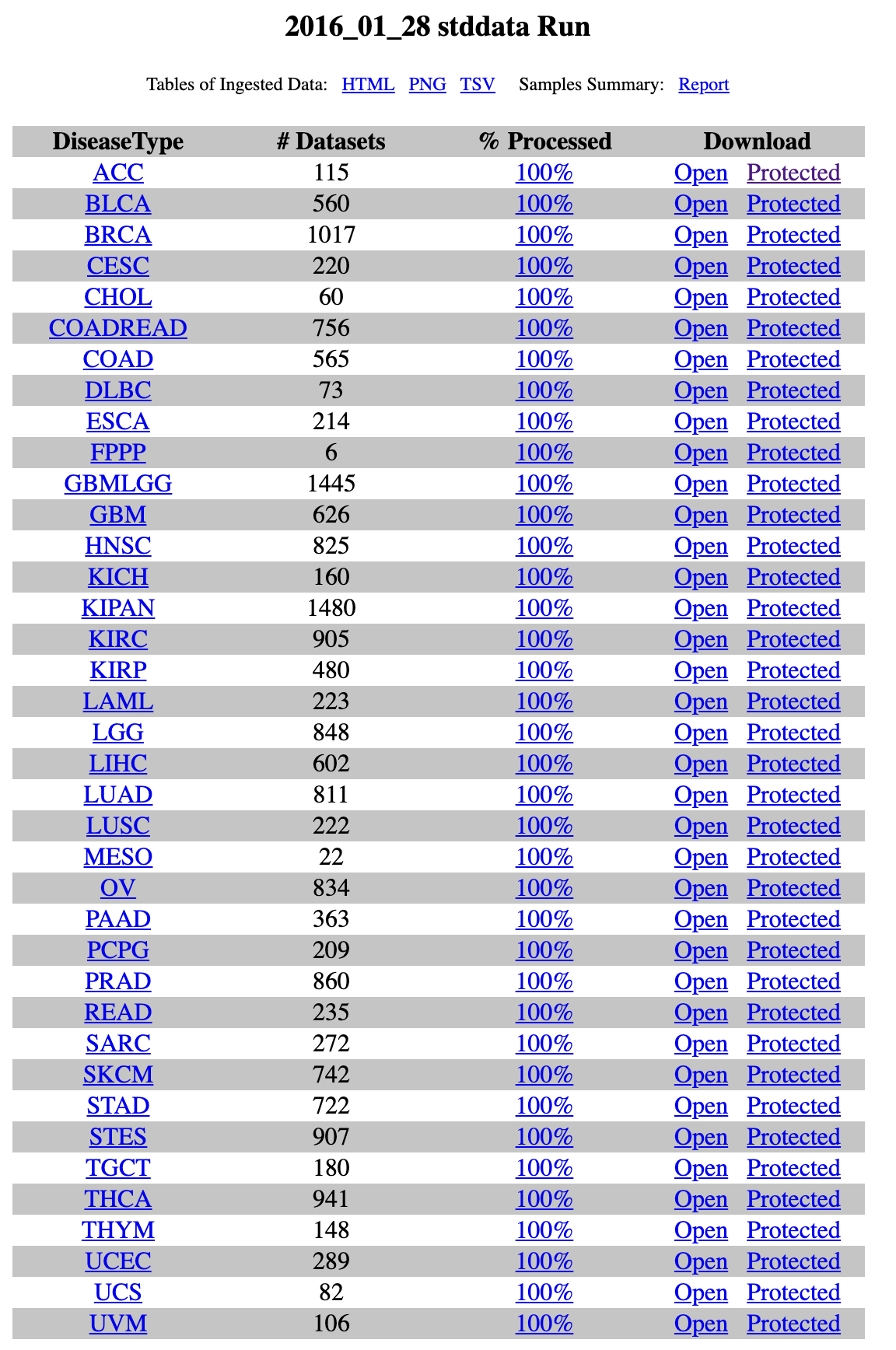
不管是哪个机构提供,都是只有分析后的数据,俗称level3数据,所以我挑选了部分,写了6个数据下载系列教程: - TCGA的28篇教程- 使用R语言的cgdsr包获取TCGA数据(cBioPortal)
- TCGA的28篇教程- 使用R语言的RTCGA包获取TCGA数据 (离线打包版本)
- TCGA的28篇教程-使用R语言的RTCGAToolbox包获取TCGA数据(FireBrowse portal)
- TCGA的28篇教程- 批量下载TCGA所有数据 ( UCSC的 XENA)
- TCGA的28篇教程-数据下载就到此为止吧
成千上万的TCGA数据挖掘文章都是围绕这些分析后的数据来的,落脚点是各种临床表型的关联分析,主要是一些统计可视化并且联系到生物学意义。pan-cancer分析
为了分析不同类型、组织起源肿瘤的共性、差异以及新课题。TCGA于2012年10月26日-27日在圣克鲁兹,加州举行的会议中发起了泛癌计划。参考:https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6000284/
其中的佼佼者是TCGA官方团队的泛癌项目 - 27论文全部是在2018年发表在Cell及其子刊上,有兴趣的可以自行查看
网址 https://www.cell.com/pb-assets/consortium/pancanceratlas/pancani3/index.html - 以及于2020年的2月5日统一发表在Nature及其子刊上的21篇文章,网址为https://www.nature.com/collections/afdejfafdb/
这里面很多数据,就是从tcga数据库过万病人的原始测序数据开始的。转录组数据
转录组数据挖掘,大家仍然是集中在mRNA,LncRNA等表达量和预后上面,但是如果你有了tcga数据库过万病人的原始测序数据,你就可以对fastq数据进行转录组的高级分析啦!
RNA编辑
指的是转录后的RNA发生的碱基插入,缺失,替换等现象,属于转录后修饰的一种,相比其他转录后修饰,比如可变剪切等,RNA编辑比较罕见,但是其作用和功能不容忽视。RNA编辑现象不仅可以发生在mRNA上,在miRNA, lncRNA等其他类型的ncRNA上也会发生。ngs技术为大规模RNA编辑位点的识别带来了便利,如果你有了tcga数据库过万病人的原始测序数据,就可以进行统一的RNA编辑位点的识别分析,不过,大概率上已经轮不到你啦:

如果你有原始的fastq测序数据,就可以走一下RNA编辑位点的识别相关分析软件,拿到结果后建立网站数据库供他人下载挖掘。可变剪切
TCGA的可变剪切也是被玩烂了的梗,大多数从一个数据库里面下载了分析好的可变剪切结果。相当于tcga数据库的新的level3数据,所以每个癌症都可以来一套同样的分析节奏。
如果你有原始的fastq测序数据,就可以走一下RNA-seq可变剪切相关分析软件,拿到结果后建立网站数据库供他人下载挖掘。
不过现在是三代测序全长转录组的时代了,以前的那些分析结果大多毫无意义,味如嚼蜡。
SnoRNA
多种RNA,包括miRNA、siRNA、piRNA、tsRNA、snRNA、snoRNA、lncRNA、circRNA等,并不是所有的都在TCGA数据库的转录组数据里面找到并且定量。
Weinberg在哺乳动物体内发现了第一个snoRNA(small nueleolar RNA,小分子核仁RNA),其主要作用是参与细胞核中前体rRNA的加工与修饰。随后在脊椎动物、酵母和植物中也发现了大量的snoRNA,它们是一类典型的ncRNA。在脊椎动物中,除少数snoRNA基因单独转录外,大部分snoRNA由蛋白质编码基因的内含子编码。酵母中除7个内含子基因和5个多顺反子snoRNA基因簇外,大部分snoRNA由单独基因编码。植物中的大部分snoRNA基因属于多顺反子基因簇,这些多顺反子基因簇部分是内含子,它们分别由2—5个snoRNA基因组成。

如果你有原始的fastq测序数据,就可以走一下SnoRNA相关分析软件,拿到结果后建立网站数据库供他人下载挖掘。融合基因
毫无疑问,已经有人挖掘并且整理好了,在数据库网页工具:https://tumorfusions.org/ 可以下载和查询针对TCGA的RNA-seq数据的全部基因融合事件,全称是:TUMOR FUSION GENE DATA PORTAL 同时还有一个:ChimerDB 4.0: an updated and expanded database of fusion genes 也提供融合基因信息。

同样的,如果你有原始的fastq测序数据,就可以走一下融合基因相关分析软件,拿到结果后建立网站数据库供他人下载挖掘。外显子数据
相比转录组数据来说,外显子数据重新挖掘的文章要少很多。更别说是重新分析原始的外显子测序数据了,我也没有时间去做系统性的调研工作了,这里就举一个例子:
微卫星不稳定
这个是从原始的外显子测序数据开始的分析,加入了新的分析软件。

现在轮到大家畅所欲言了
给你tcga数据库过万病人的原始测序数据你可以做什么?
大家可以发挥自己的生物学背景优势,畅所欲言,如果是做免疫的,可以考虑从RNA-seq里面分析免疫组库相关基因表达量,有点类似于m6A相关基因或者自噬相关基因的数据挖掘分析:
文末友情宣传
强烈建议你推荐我们生信技能树给身边的博士后以及年轻生物学PI,帮助他们多一点数据认知,让科研更上一个台阶:
- 生信爆款入门-全球听(买一得五)(第4期),你的生物信息学入门课
- 数据挖掘第2期(两天变三周,实力加量),医学生/临床医师首选技能提高课
- 生信技能树的2019年终总结 ,你的生物信息学成长宝藏
- 2020学习主旋律,B站74小时免费教学视频为你领路,还等什么,看啊!!!