大部分的生物学高通量数据处理后都是得到基因集,不管是上调下调表达基因集,还是共表达的模块基因集,都是需要注释到生物学功能数据库来看基因集的意义,最常见的是GO/KEGG数据库啦,还有很多其它在MsigDB的,比如reactome和biocarta数据库等等。
这样分析起来就很麻烦,尤其是GO数据库,还有BP,CC,MF的区别,这个时候推荐使用Y叔的神器:
library(ggplot2)
library(stringr)
library(clusterProfiler)
# 我这里演示的是brown_down_gene,是WGCNA的一个模块,基因集
# 因为表达矩阵是symbol,所以需要转为ENTREZID,才能走clusterProfiler的函数。
gene.df <- bitr(brown_down_gene$symbol, fromType="SYMBOL",
toType="ENTREZID",
OrgDb = "org.Hs.eg.db")
go <- enrichGO(gene = gene.df$ENTREZID, OrgDb = "org.Hs.eg.db", ont="all")
barplot(go, split="ONTOLOGY")+ facet_grid(ONTOLOGY~., scale="free")
会得到如下所示的图,当然,理解起来需要耗费一点功夫,如果你是第一次看到的话!不仅仅是要理解GO数据库,以及BP,MF,CC的分类系统,超几何分布检验,不同的阈值过滤,筛选指标等等。
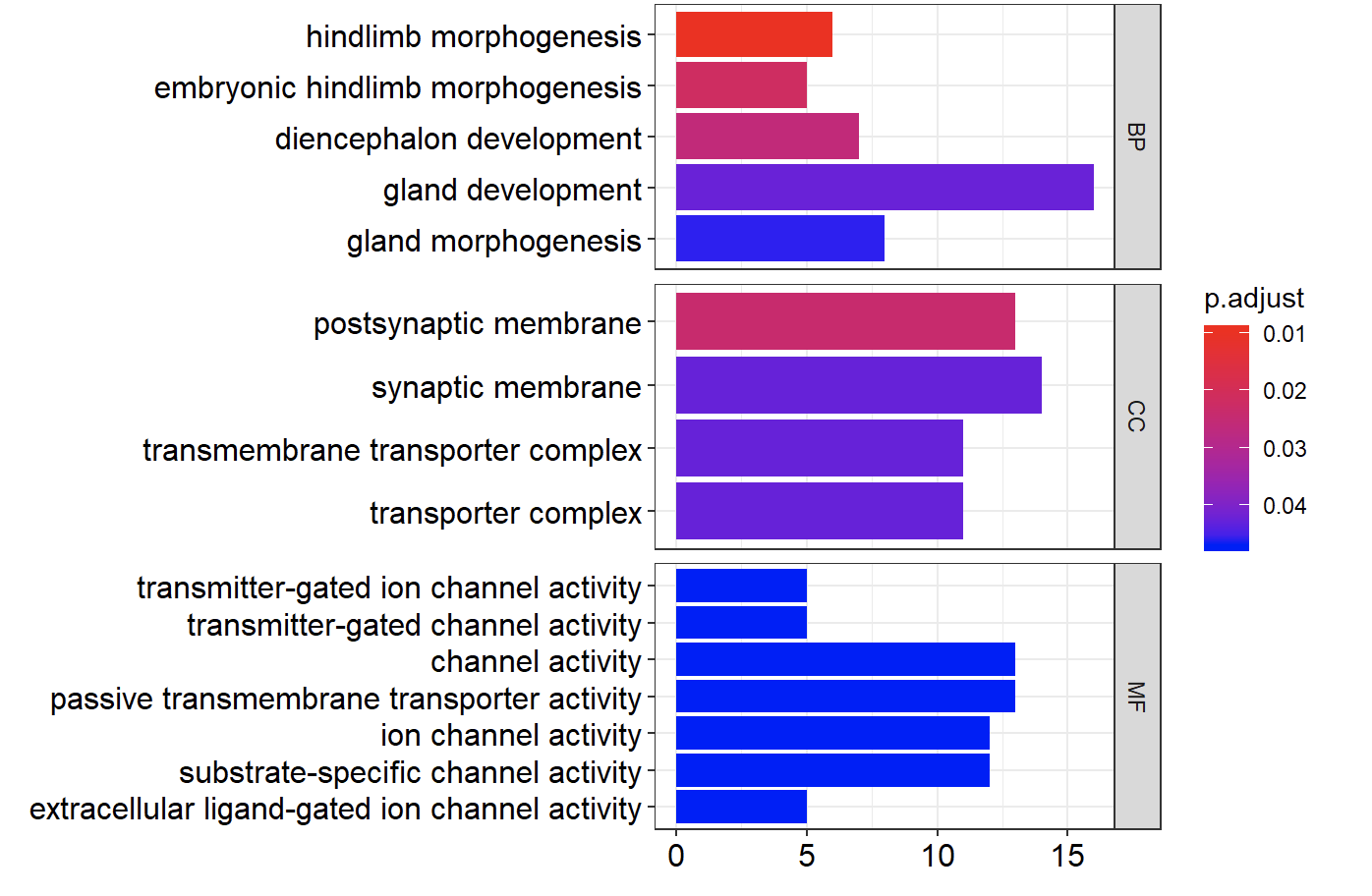
因为上面的代码并没有修改默认的统计学指标筛选参数,如果你的基因确实没有规则,有可能拿不到结果哦!这个时候可以设置: pvalueCutoff = 0.9, qvalueCutoff =0.9 甚至为1,来不做筛选。而且基因集的大小也是被限制了。
如果你想分开计算上下调基因的GO数据库注释
而且还想保留富集分析结果到csv文件,代码如下:
library(ggplot2)
library(stringr)
library(clusterProfiler)
# 通过前面的差异分析,我们拿到了 gene_up 和 gene_down 这两个基因集
# 后面的分析,只需要 gene_up 和 gene_down 这两个变量即可
go_up <- enrichGO(gene_up,
OrgDb = "org.Hs.eg.db",
ont="all",
pvalueCutoff = 0.9,
qvalueCutoff =0.9)
go_up=DOSE::setReadable(go_up, OrgDb='org.Hs.eg.db',keyType='ENTREZID')
write.csv(go_up@result,paste0(pro,'_go_down.up.csv'))
barplot(go_up, split="ONTOLOGY",font.size =10)+
facet_grid(ONTOLOGY~., scale="free") +
scale_x_discrete(labels=function(x) str_wrap(x, width=50))+
ggsave(paste0(pro,'gene_up_GO_all_barplot.png'))
go_down <- enrichGO(gene_down,
OrgDb = "org.Hs.eg.db",
ont="all",
pvalueCutoff = 0.9,
qvalueCutoff =0.9)
go_down=DOSE::setReadable(go_down, OrgDb='org.Hs.eg.db',keyType='ENTREZID')
write.csv(go_down@result,paste0(pro,'_go_down.up.csv'))
barplot(go_down, split="ONTOLOGY",font.size =10)+
facet_grid(ONTOLOGY~., scale="free") +
scale_x_discrete(labels=function(x) str_wrap(x, width=50))+
ggsave(paste0(pro,'gene_down_GO_all_barplot.png'))
其实就是两个独立的基因集,独立的走enrichGO流程啦!
多组基因集的KEGG数据库富集
有趣的是,如果你是多组基因,不仅仅是上下调,甚至可以走compareCluster流程,不过Y叔的这个函数总是喜欢在线获取KEGG数据库的最新信息,这一点对很多人来说,考验网速:
# 这里需要制作一个 DEG 的数据框,其中有两列ENTREZID,是基因id,和new是分组信息
xx.formula <- compareCluster(ENTREZID~new, data=DEG, fun='enrichKEGG')
dotplot(xx.formula, x=~GeneRatio) + facet_grid(~new)
如果是多组基因集走GO数据库富集
如下,构建一个数据框,list_de_gene_clusters, 含有两列信息:
list_de_gene_clusters <- split(de_gene_clusters$ENTREZID,
de_gene_clusters$cluster)
# Run full GO enrichment test
formula_res <- compareCluster(
ENTREZID~cluster,
data=de_gene_clusters,
fun="enrichGO",
OrgDb="org.Mm.eg.db",
ont = "BP",
pAdjustMethod = "BH",
pvalueCutoff = 0.01,
qvalueCutoff = 0.05
)
# Run GO enrichment test and merge terms
# that are close to each other to remove result redundancy
lineage1_ego <- simplify(
formula_res,
cutoff=0.5,
by="p.adjust",
select_fun=min
)
感兴趣的可以把这个结果跟3个出名的网页工具进行比较
另外,强推Y叔clusterProfiler的一些可视化方法
可视化方法函数列表:
- barplot
- cnetplot
- dotplot
- emapplot
- gseaplot
- goplot
- upsetplot
好几个都是以前没有介绍过的,有趣的是我准备浏览这些可视化函数的帮助文档的时候,看到了这样的话:
重点来了,Y叔特意为其包写了一本书来介绍其用法。Please go to https://yulab-smu.github.io/clusterProfiler-book/ for the full vignette.
文末友情宣传
强烈建议你推荐我们生信技能树给身边的博士后以及年轻生物学PI,帮助他们多一点数据认知,让科研更上一个台阶:
- 生信爆款入门-全球听(买一得五)(第4期),你的生物信息学入门课
- 数据挖掘第2期(两天变三周,实力加量),医学生/临床医师首选技能提高课
- 生信技能树的2019年终总结 ,你的生物信息学成长宝藏
- 2020学习主旋律,B站74小时免费教学视频为你领路,还等什么,看啊!!!