生意这样的商业活动,理论上可以激励创造,让参与交易的双方都受益,才有可能持续。比如你不可能花费半年时间去系统性学习R语言和Linux操作,处理fastq的单细胞测序数据,做统计可视化图表,就为了一辈子的一个项目。所以理论上你的最优解决方案是委托专业的生信工程师,比如我们就在单细胞天地发布过:[单细胞转录组下游分析这样报价合理吗?](https://mp.weixin.qq.com/s/K0Eysl3mwBsqFMKT9SCe_w) ,试图促进大家合作,可惜的是虽然几万人参与讨论,但是最后却不欢而散。专业的工程师觉得为客户学习一个R包收费2000合情合理,但是委托者觉得一个项目全套分析收2000才合理。
正好,最近我的学徒也分享了他在分析他们课题组数据项目的一个感悟,值得分享给大家,大家可以思考一下,为什么会有这样的生意,交易双方都觉得血亏呢?
起初是求助一段perl代码
学徒在文献里面找到了下面这段perl代码,不太看得懂,根据大致猜测,翻译成了后面这段R代码,请曾老师帮忙看一下猜得对不对:
# calculate a discrete logarithmically-stepped integer index from RPKM values
INDEX_FUNCTION='function getIndex(rpkm){ \
ff=log('MAX_RPKM'/'MIN_PRKM') \
i=rpkm/'$MIN_RPKM'; \
i=i<1?0:i; \
i=i>xrpkm?xrpkm:i; \
i=i>0?log(i)/log(ff):-1; \
i=int(i)+1; \
return i; \
}'
getIndex(rpkm)=function(rpkm){
i=rpkm/MIN_RPKM
ff=log(MAX_RPKM/MIN_PRKM)
if(i<1){i=0}
if(i>xrpkm){i=xrpkm}
if(i>0) {log(i)/log(ff)=log(i)/log(ff)-1} #化简之后相当于i=rpkm/MIN_RPKM/ff
i=floor(i)+1
return(i)
}
其实,如果不是擅长这两种编程语言,这两个代码看起来都是天书。
我还没有来得及帮忙解决这个问题,第二天,学徒主动发给我了他的解决方案!
是隐马尔可夫模型划分基因组单元
昨天的perl代码看懂了,今天来把坑填上。
- 昨天的问题来自一个叫 segment.pl 的软件,用来将连续的基因组划分成离散的片段。划分的依据是用户给定的一些特征,比如新生RNA转录情况,或者CNV。
- perl代码:
# calculate a discrete logarithmically-stepped integer index from RPKM values
MIN_RPKM=0.001 # the minimum RPKM value; bins with RPKM<$MIN_RPKM all have index=0
MAX_RPKM=100 # the maximum RPKM value; bins with RPKM>$MAX_RPKM all have the same maximum index
OBS_FOLD_FACTOR=2 # observation indices stepped logarithmically every $OBS_FOLD_FACTOR-fold from $MIN_RPKM to $MAX_RPKM
#以上这些是软件文档里的默认参数和解释
INDEX_FUNCTION='function getIndex(rpkm){ \
xrpkm='$MAX_RPKM'/'$MIN_RPKM'
ff=OBS_FOLD_FACTOR
i=rpkm/'$MIN_RPKM'; \
i=i<1?0:i; \ #若i<1,则i=0;否则i=i,即不变
i=i>xrpkm?xrpkm:i; \ #若i>xrpkm,则i=xrpkm;否则不变
i=i>0?log(i)/log(ff):-1; \ #若i>0,则i=log(i)/log(ff);否则i=-1
i=int(i)+1; \ #i向下取整后加1
return i; \
}'
?代表判断,若为真,则取:前的值;若为假,则取:后的值
- 翻译成R:
MIN_RPKM=0.001
MAX_RPKM=100
OBS_FOLD_FACTOR=2
getIndex=function(rpkm){
xrpkm=MAX_RPKM/MIN_RPKM
ff=OBS_FOLD_FACTOR
i=rpkm/MIN_RPKM
if(i<1){i=0}
if(i>xrpkm){i=xrpkm}
if(i>0) {i=log(i,base=ff)}
else {i=-1}
i=floor(i)+1
return(i)
}
代码作用
-
我花了九牛二虎之力终于明白这段代码的作用了:
-
(在这之前已经把基因组划分成了1kb的bin,计算了每个bin的nascent RNA表达量RPKM)
- 将每个bin的RPKM换算成index,其换算方法为:若RPKM低于设定的下界,则index=0;若RPKM高于设定的上界,则令RPKM=设定的上界
- 然后对于RPKM不低于下界的bin,做以下计算:先将RPKM处以设定的下界,然后以ff变量为底取对数,再向下取整后加1
- 用人话翻译一下:对在一定范围内的表达量,取对数后进行等分,从而将表达量转为index(正整数);表达量超过范围时,将index设定为index的最小或最大值。
-
原文是这么描述这段代码的作用的:
Corrected bin data were then indexed into a series of bounded integer observation values from 0 through 17, logarithmically spaced across bin RPKM values from <0.0005 to >100.
- 以及这么描述的:
Score the bins by rounding each into one of 17 logarithmically distributed RPKM input states.
- 想通了以后觉得作者这么写也没问题,但是对于不懂的人来说就是天书啊orz
故事背景——用隐马尔可夫模型对基因组进行分割
上面这段代码看起来已经挺复杂了,但它其实只是整篇文章的一个小插曲。如果有兴趣的话,可以看看下面的故事背景:
目的:切割基因组
- 需求:将连续的基因组分割成离散的转录单元。
- 输入:全基因组每个1kb的bin的RPKM
- 核心:隐马尔可夫模型(等下讲)
- 直接输出:每个bin的转录状态,即index(有限范围的正整数)。
- 间接输出:index高于阈值的bin认为是转录的,index低于阈值认为是不转录的
- 最终输出:相邻的转录的bin连接起来,构成转录单元(transcription unit)。这是nascent RNA测序中一个重要的概念。
整个workflow就是以上这么回事,难点在于隐马尔可夫模型(HMM),也就是如何根据我们测到的RPKM,来判断一个bin有没有表达。
(我内心:这不是直接划个阈值就完事了吗???好吧,存在即合理,简单地划个阈值肯定会有诸多问题的)
下面我尽可能通俗地讲一下HMM是怎么回事:
做法:隐马尔可夫模型
- 隐马尔可夫模型的四要素:真实状态(state),观察到的状态(observed value),发射概率(emission possibility),转换概率(transition possibility)
- 对于nascent RNA来说,测序测到的表达量是observed value,而我们想知道的转录单元是state。为什么要用HMM?是因为测序(observed value)不可能完完全全准确地反映真实状态(state),所以要用HMM来帮助我们判断真实状态。
- 发射概率—— 在某一种真实状态下,产生某一种观察状态的概率,它是一个条件概率:P ( value=j | state=i )。在我们这个问题中,发射概率是这样算的:
- 先把每个bin都注释到唯一的基因(如果没注释到,或者注释到多个基因,则舍去这个bin)。
- 同时把每个bin和每个gene的RPKM都转换成index(也就是开头的那段perl代码了)
- 然后计算index=j的bin落在index=i的基因里的频率。i和j都遍历各自的所有可能,然后就会得到一个条件概率矩阵,这个矩阵就是emission possibility file。

-
转换概率—— 从上一个单位到下一个单位时(在这里就是上一个bin到下一个bin),真实状态从a变成b(也就是转录状态发生改变)的概率。作者设定了转换概率为0.005,也就是说一个转录本有0.995的概率会继续转录下去,有0.005的概率会终止。反之,一个不转录的区域有0.005的概率会开始转录。
-
四大要素都准备好了之后,就可以用segment软件来计算了。(至于其中的算法,用的是Viterbi,应该只有理工科学生才会去关注了)
cat $input | segment -e $EMISS_PROB_FILE -z 0.5 -p 0.995 > segment_output.txt
# input文件中是每个bin的信息,包含三列,分别是index,chr,起始位置
# EMISS_PROB_FILE中是发射概率矩阵
# -z 0.5 代表全基因组的bin中,假设不转录的bin占50%
# -p 0.995 代表转换概率为0.005
以上就是隐马尔可夫模型的故事。我相信它这么复杂,没有几个人会用它来研究转录问题的。
所以做nascent RNA和transcription unit研究的人也不是很多,国内应该也没有公司会做这个测序。
毕竟吃力不讨好,看懂这个模型花了我将近两周时间,写这个文档来分享都花了一个小时,谁乐意跳这个大坑呢?
你现在觉得专业的工程师为了你的课题帮忙看一个算法看一个R包或者perl代码,收费多少合适呢?
你以为这样就完了吗?实际上还有进阶资料,关于HMM计算转录单元,不过,实在是太冷门了,如果你确实不从事这方面就不需要再看了哈。
HMM计算转录单元进阶资料
- 利用segment软件计算完后,得到的是每个bin的新的index
- 把相邻的且index相同的bin合并成segment
- 把index<=3的segment过滤掉,留下的segment就认为是转录单元了(transcription unit)
- 在检查了结果之后发现,index=15(这个数据集里最大的index)的转录单元存在显著的假阳性,所以我们把15也给过滤了。
最终效果如下:
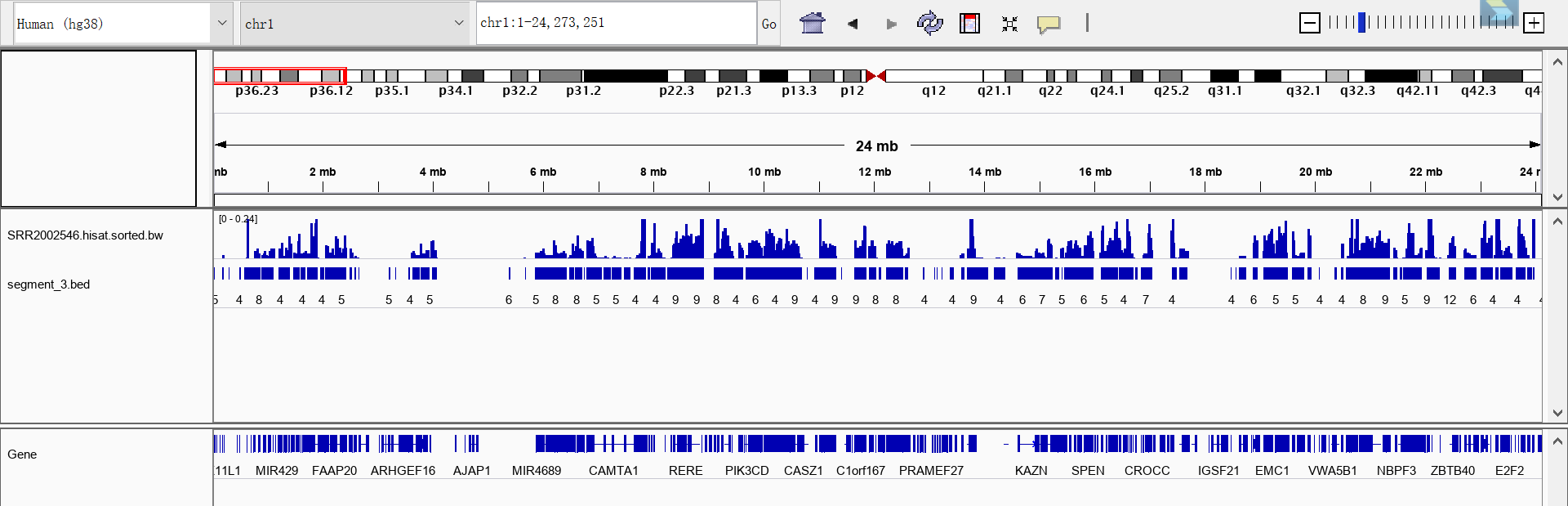
第一行是nascent RNA测序的bw文件,第二行是我们计算的transcription unit的bed文件,下面的数字就是每个转录单元的index,最下面一行是IGV自带的基因注释。
可以发现,转录单元与bw是能很好地吻合的。图中有一些小的转录单元的bw看起来没有信号,但放大之后看其实是有的,只是没有那么明显而已。看来这个算法还不错,能够比较好地反映转录情况。
另外,我们把transcription unit和IGV的基因注释比较一下可以看到,它们在挺多地方都是存在区别的。也就是说如果我们以转录单元为单位来计算表达量,会比以基因为单位来计算更加准确。所以,虽然用隐马尔可夫模型来计算转录单元非常麻烦,但是这么做是值得的,我收回前天最后一段的吐槽。
参考文献:
-
Use of Bru-Seq and BruChase-Seq for genome-wide assessment of the synthesis and stability of RNA. Methods
-
Coordinated regulation of synthesis and stability of RNA during the acute TNF-induced proinflammatory response. PNAS
-
Large transcription units unify copy number variants and common fragile sites arising under replication stress. Genome Research
- Intragenic origins due to short G1 phases underlie oncogene-induced DNA replication stress. Nature
- DNA repair and recovery of RNA synthesis following exposure to ultraviolet light are delayed in long genes. Nuc Acid Res
文末友情宣传
强烈建议你推荐给身边的博士后以及年轻生物学PI,多一点数据认知,让他们的科研上一个台阶:
- 全国巡讲全球听(买一得五) ,你的生物信息学入门课
- 生信技能树的2019年终总结 ,你的生物信息学成长宝藏
- 2020学习主旋律,B站74小时免费教学视频为你领路