最近看到很多人讨论基于nextflow的nf-core,里面存储了几十种NGS组学数据分析流程哦,而且文章发表在NBT。最早应该是生信技能树的学习大使《二货潜》发在朋友圈的,他同时也推荐了 加拿大生物信息学研讨会资源宝藏 。然后我看到生信技能树的宝藏男孩《唐医生》在 [菜鸟团一周文献推荐(No.50)](https://mp.weixin.qq.com/s/4VGvBuuJkxeDb-cLaabQrg) ,再次提到多种组学的生信分析流程大整合:
文章信息
题目:The nf-core framework for community-curated bioinformatics pipelines
杂志:Nature Biotechnology
时间:Published: 13 February 2020
链接: https://www.nature.com/articles/s41587-020-0439-x
目前 nf-core 共包含了 27 种分析流程,如下:
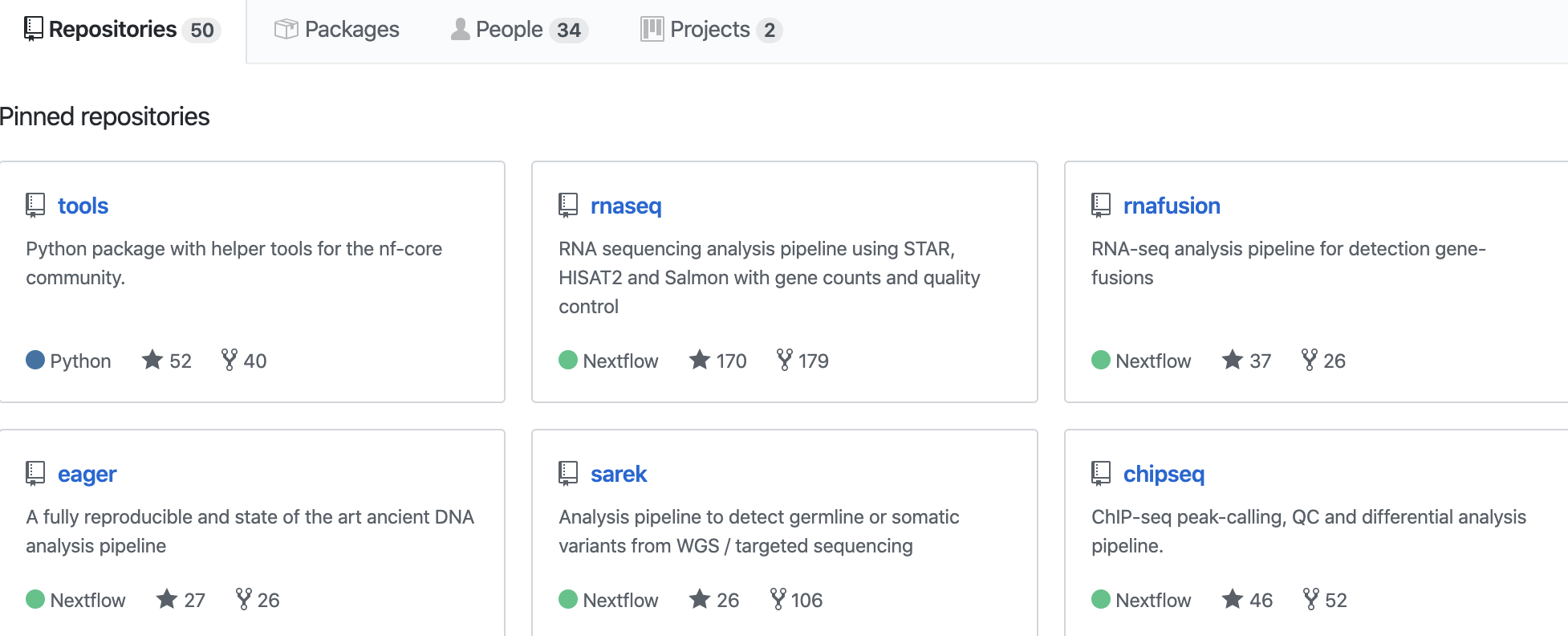
而nf-core里面的不同流程,本质上就是一些测试数据和写好的配置文件,方便我们的nextflow调用配置文件来处理测序数据,每一种流程都是一些数据的处理步骤的集合!
首先需要安装nextflow
装方法参考这个链接:https://nf-co.re/usage/installation
首先需要检查 java 版本,大于 8
# Make sure that Java v8+ is installed:
java -version
openjdk version "1.8.0_242"
OpenJDK Runtime Environment (build 1.8.0_242-8u242-b08-0ubuntu3~18.04-b08)
OpenJDK 64-Bit Server VM (build 25.242-b08, mixed mode)
然后下载及安装 Nextflow,需要点时间(在中国大陆访问速度很慢,建议放弃这个策略)
# Install Nextflow
curl -fsSL get.nextflow.io | bash
# 然后添加到环境变量
或者也可以用 conda 安装(推荐,因为可以设置镜像)
# 先安装conda并且配置镜像
wget -c https://mirrors.tuna.tsinghua.edu.cn/anaconda/miniconda/Miniconda3-latest-Linux-x86_64.sh
bash Miniconda3-latest-Linux-x86_64.sh # (一系列互动设置)
source ~/.bashrc # (安装好的conda必须启动)
conda config --add channels https://mirrors.tuna.tsinghua.edu.cn/anaconda/cloud/bioconda/
conda config --add channels https://mirrors.tuna.tsinghua.edu.cn/anaconda/cloud/conda-forge/
conda config --add channels https://mirrors.tuna.tsinghua.edu.cn/anaconda/pkgs/free/
conda config --add channels https://mirrors.tuna.tsinghua.edu.cn/anaconda/pkgs/main/
conda config --set show_channel_urls yes
# 可以创建创建新环境,单独存放 nextflow 程序
conda create -n nf-core -y
conda activate nf-core
# 然后安装 nextflow
conda install -y nextflow
假如需要更新,就运行 nextflow self-update 或 conda update nextflow
本地运行nextflow
我们这里测试一下nf-core里面的RNAseq的流程吧,参考github:https://github.com/nf-core/rnaseq/ (PS : 因为是GitHub上面的东西,同样的考验在中国大陆的网速)
nextflow run nf-core/rnaseq
需要说明的是,该工具会自己 下载GitHub里面的 nf-core/rnaseq 资料,然后走一些他们预设好的分析步骤:
[- ] process > get_software_versions -
[- ] process > get_software_versions -
[- ] process > makeBED12 -
[- ] process > makeSTARindex -
[- ] process > fastqc -
[- ] process > trim_galore -
[- ] process > star -
[- ] process > rseqc -
[- ] process > preseq -
[- ] process > markDuplicates -
[- ] process > qualimap -
[- ] process > dupradar -
[- ] process > featureCounts -
[- ] process > merge_featureCounts -
[- ] process > stringtieFPKM -
[- ] process > sample_correlation -
[- ] process > multiqc -
[- ] process > output_documentation -
第一次使用一个流程,会调用nf-core/rnaseq/environment.yml 文件配置一个独立的conda环境(nf-core-rnaseq),安装一系列软件,如下:
# You can use this file to create a conda environment for this pipeline:
# conda env create -f environment.yml
name: nf-core-rnaseq-1.4.2
channels:
- conda-forge
- bioconda
- defaults
dependencies:
## conda-forge packages, sorting now alphabetically, without the channel prefix!
- matplotlib=3.0.3 # Current 3.1.0 build incompatible with multiqc=1.7
- r-base=3.6.1
- conda-forge::r-data.table=1.12.4
- conda-forge::r-gplots=3.0.1.1
- conda-forge::r-markdown=1.1
## bioconda packages, see above
- bioconductor-dupradar=1.14.0
- bioconductor-edger=3.26.5
- bioconductor-tximeta=1.2.2
- bioconductor-summarizedexperiment=1.14.0
- deeptools=3.3.1
- fastqc=0.11.8
- gffread=0.11.4
- hisat2=2.1.0
- multiqc=1.7
- picard=2.21.1
- preseq=2.0.3
- qualimap=2.2.2c
- rseqc=3.0.1
- salmon=0.14.2
- samtools=1.9
- sortmerna=2.1b # for metatranscriptomics
- star=2.6.1d # Don't upgrade me - 2.7X indices incompatible with iGenomes.
- stringtie=2.0
- subread=1.6.4
- trim-galore=0.6.4
是不是get到了conda的一个高级用法!conda创建环境以及安装软件,本质上是新建了一个文件夹,下载了一些文件而已,每个流程涉及到的软件文件多达几个G,都在work目录。
如果运行成功,会在工作目录生成几个文件夹:
work # Directory containing the nextflow working files
results # Finished results (configurable, see below)
.nextflow_log # Log file from Nextflow
# Other nextflow hidden files, eg. history of pipeline runs and old logs.
实际运行时也是一行代码,对自己的样本的测序数据,指定好fq文件,以及参考基因组即可:
nextflow run nf-core/rnaseq --reads 'path/to/data/sample_*_{1,2}.fastq' --genome GRCh38 -profile conda
需要注意的是,fq 文件的路径需要加上一对引号 ,同一样本的 fq1 和 fq2 用 * 来匹配,fq 文件支持 gz 压缩格式。避免重复调用conda创建环境以及安装软件,保证每个流程在同一个目录下运行比较好。
不过可能需要适当修改一下流程,比如参考基因组,可以在 pull 下来的仓库的 rnaseq/conf/igenomes.config 查看到,比如人类的就有 GRCh37 ,GRCh38,hg19,hg38
所有的参考基因组如下:
'GRCh37' ,'GRCh38' ,'GRCm38' ,'TAIR10' ,'EB2' ,'UMD3.1' ,'WBcel235' ,'CanFam3.1' ,'GRCz10' ,'BDGP6' ,'EquCab2' ,'EB1' ,'Galgal4' ,'Gm01' ,'Mmul_1' ,'IRGSP-1.0' ,'CHIMP2.1.4' ,'Rnor_6.0' ,'R64-1-1' ,'EF2' ,'Sbi1' ,'Sscrofa10.2' ,'AGPv3' ,'hg38' ,'hg19' ,'mm10' ,'bosTau8' ,'ce10' ,'canFam3' ,'danRer10' ,'dm6' ,'equCab2' ,'galGal4' ,'panTro4' ,'rn6' ,'sacCer3' ,'susScr3'
如果使用的是其他物种,需要在上面文件中以下面的格式添加相关信息:
params {
genomes {
'GRCh37' {
star = '<path to the star index folder>'
fasta = '<path to the genome fasta file>' // Used if no star index given
gtf = '<path to the genome gtf file>'
bed12 = '<path to the genome bed file>' // Generated from GTF if not given
}
// Any number of additional genomes, key is used with --genome
}
}
或者在实际运行时加上各种参数:
--star_index '/path/to/STAR/index' \
--hisat2_index '/path/to/HISAT2/index' \
--fasta '/path/to/reference.fasta' \
--gtf '/path/to/gene_annotation.gtf' \
--gff '/path/to/gene_annotation.gff' \
--bed12 '/path/to/gene_annotation.bed'
如果要对流程进行修改,比如有些人可能计算资源不够,跑人类数据 star 这一步会比较感人,内存要大于 38G 才能跑得动。可以指定用 hisat2 进行比对就好,加上参数 --aligner hisat2 或者配置文件 .nextflow/assets/nf-core/rnaseq/nextflow.config 中设置 params.aligner = 'hisat2'
更多的设置见:https://github.com/nf-core/rnaseq/blob/master/docs/usage.md
实际上,每个参数,都可以在运行这个流程的时候传入,但是不动了流程里面的每个步骤每个软件每个参数,你也就不从修改起。
其实这些技术流程的视频教程在好几年前,我就全部免费共享在b站,如果你没有看,说明你可能并不值得培养,加入人家团队也很勉强。而且我同步分享了视频配套讲义和教辅材料:
- 学徒第1月,基础知识介绍掌握:文档链接:https://mubu.com/doc/38tEycfrQg 密码:vl3q
- 学徒第2月,RNA-seq数据分析实战训练:文档链接:https://mubu.com/doc/38y7pmgzLg 密码:p6fo
- 学徒第3月,WES数据分析实战训练:文档链接:https://mubu.com/doc/1iDucLlG5g 密码:7uch
- 学徒第4月,ChIP-seq数据分析实战训练:文档链接:https://mubu.com/doc/11taEb9ZYg 密码:wk29
也为每个组学视频课程,设置了练习题,不知道大家是否有学习呢?
基本上每个过来我这边学习一个月以上的学徒我都会让他们学习多种组学(围绕着中心法则),而且有了Linux基础和R语言能力后, 跟着我们的视频教程很容易就学会基础流程,毫无压力。
基于 docker 或 Singularity 来运行nextflow
这个稍微有点复杂,我还没有测试,
我在生信技能树上面写过部分docker教程, 目录如下:
- 用集成了anaconda的docker快速布置生信分析平台
- 我学会docker啦!希望你也可以学会
- 跟着jimmy学docker系列之第2讲:一个软件一个容器
- 跟着jimmy学docker系列之第3讲:为何不创建自己的docker容器呢?
- 跟着jimmy学docker系列之第4讲:docker容器资源调度问题(MAC版本)
- 使用阿里云+Docker分析RNA-Seq与ChIP-Seq
- Docker应用之一键化安装Wordpress(无需代码基础)
- 如何从看不懂Dockerfile到创建自己的镜像
用的不多,就不浪费时间测试了。
参考:
- 生信框架学习-nextflow, 徐州更 生信媛 2017-12-04
- 初识Nextflow (系列之一) , CJchen 生信札记 2018-11-02
文末友情宣传
强烈建议你推荐给身边的博士后以及年轻生物学PI,多一点数据认知,让他们的科研上一个台阶:
- 全国巡讲全球听(买一得五) ,你的生物信息学入门课
- 生信技能树的2019年终总结 ,你的生物信息学成长宝藏
- 2020学习主旋律,B站74小时免费教学视频为你领路