昨天我们在生信技能树解读了 从招聘信息看一个合格的生信工程师该会哪些 ,朋友圈有一小撮人冷嘲热讽,说辛辛苦苦学了那么多,工资也就万把块钱每个月。我就呵呵,谁让你当年选择了生物呢?本来应该是去卖保险信用卡股票期货的你,因为遇到了生信,有幸找到一份养家糊口的工作你还得寸进尺?好不容易你才走入了社会正常运转的一环,成为一个不可或缺的螺丝钉,对没有背景的你来说,已经是鲤跃龙门啦!
我不知道到底多少钱的工资才符合你的期望,社会上反正多的是一夜暴富,收入显赫的职业,你做不来,在我们生信技能树这里抱怨是没有用的。但是,我仍然是可以给你秀一下,学生物信息学数据分析,也不是不可以挣钱。记住,是挣钱,合理的挣钱,不是赚钱!
话说,也许是五年前,或者是四年前,我还在写生信菜鸟团博客的时候,分享了大量的RNA-seq分析实战教程,非常的受欢迎。遇到一个粉丝,跟着我的教程学了好久,至少邮件问了我不下十个问题,最后还是搞不定。其实就是因为没有学Linux,自己的Windows电脑搞虚拟机折腾起来累人。然后博士毕业需要用那个数据,确实没办法,就干脆找我,开价800块钱,我帮忙拿到表达矩阵。
公共数据
-
GEO地址是:https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE35005
-
SRA地址是:https://trace.ncbi.nlm.nih.gov/Traces/sra?study=SRP010262
首先下载sra文件
内容如下:
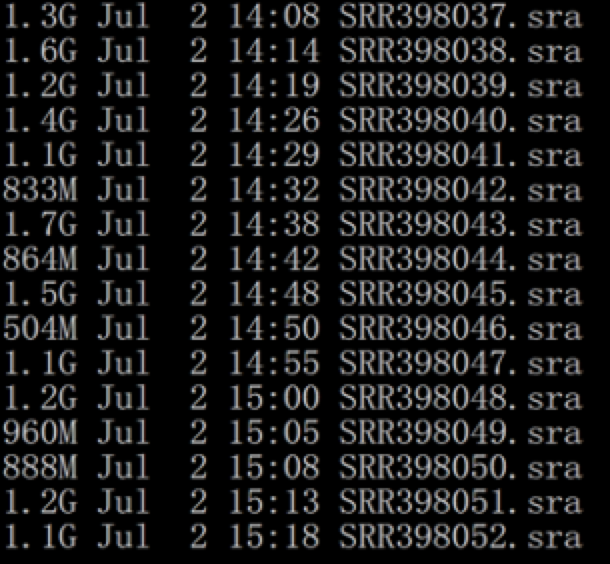
然后转为fastq文件
可以看到,都是单端测序数据,如下:
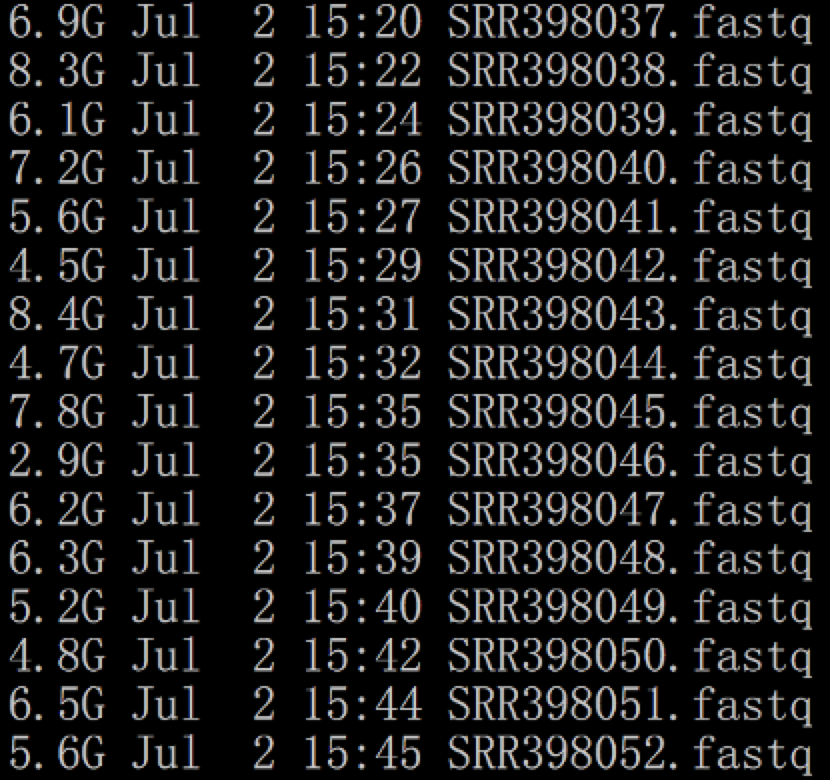
这些fastq文件需要根据项目信息来合并,因为一个样本测到了两个fastq文件。如下:
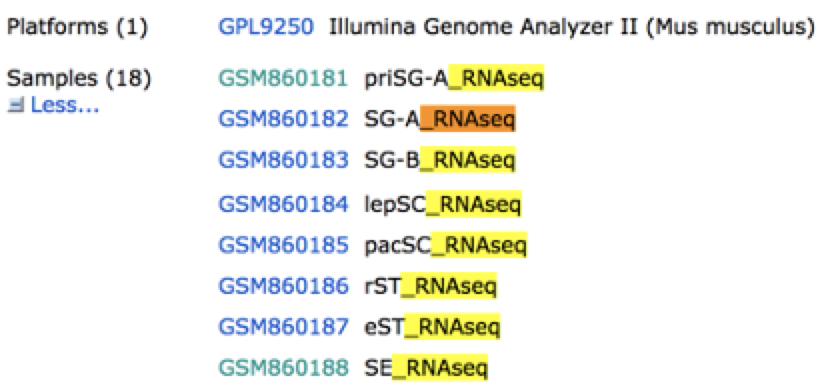
接着走RNA-seq的上游分析流程
主要是拿到bam文件和表达矩阵
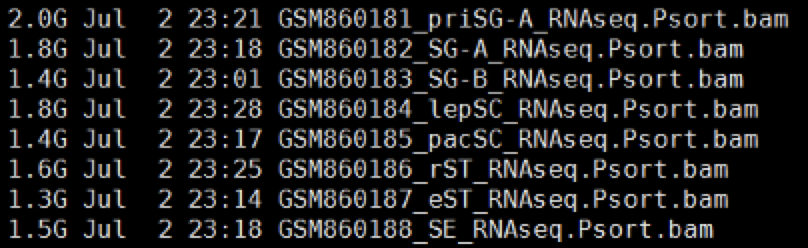
流程代码,真心很简单,但是是对会的人来说,这个ngs上游分析能力,真的很简单,但是如果你并没有学过Linux的shell编程,下面代码的确是难如天书!
# for i in {0..5};do bash step1-fastp-hisat2-se.sh `pwd` config.se 6 $i;done
analysis_dir=$1
config_file=$2
number1=$3
number2=$4
conda activate rna
index=/home/yb77613/reference/index/hisat/hg38/genome
# for config.se
# sampleName and fastq files(for single end)
# SCBO-9_orgP9 /home/bladder/1.1.raw_fq/SCBO-9_orgP9.fastq.gz
cat $config_file |while read id
do
echo $id
arr=($id)
fq=${arr[1]}
sample=${arr[0]}
if((i%$number1==$number2))
then
## step1: basic QC for fastq files
if [ ! -f ok.fastp.$sample.status ]; then
fastp -i $fq -o 2.1.clean_fq/${sample}.fq.gz \
--thread=4 --length_required=36 --n_base_limit=6 \
--compression=6 -R ${sample} \
1>0.0.logs/log.fastp.${sample}.txt 2>&1
fi ## end for if files
if [ $? -eq 0 ]; then
touch ok.fastp.$sample.status
else
echo "fastp failed" $sample
fi
fastqc $fq -O 0.1.qc
fastqc 2.1.clean_fq/${sample}.fq.gz -O 0.1.qc
## step2: alignment by hisat2
if [ ! -f ok.hisat2.$sample.status ]; then
hisat2 -p 4 -x $index -U 2.1.clean_fq/${sample}.fq.gz | \
samtools sort -O bam -@ 4 -o - > 3.1.hisat2_align/${sample}.se.bam
fi ## end for if files
if [ $? -eq 0 ]; then
touch ok.hisat2.$sample.status
else
echo "hisat2 failed" $sample
fi
fi # check the number1 number2
i=$((i+1))
done
对所有的bam文件,统一进行定量流程
代码都是三五年前就写好的:
gtf="/home/reference/gtf/gencode/gencode.v32.annotation.gtf"
bin_featureCounts="/home/biosoft/featureCounts/subread-1.5.3-Linux-x86_64/bin/featureCounts";
$bin_featureCounts -T 4 -t exon -g gene_name -a $gtf -o all.counts.id.txt ../hisat2/*.bam 1>counts.id.log 2>&1
有了表达矩阵就走一个差异分析咯
我也不记得我分享过多少次代码,多到大家都责怪我在炒冷饭了。
RNA-seq我们在生信技能树应该是至少推出了400篇教程,而且是我们全国巡讲的标准品知识点,其中还有一个阅读量过两万的综述翻译及其细节知识点的补充:
相信大家听完了我B站的RNA-seq分析流程后,对这个数据的应用方向都不陌生。
简化一下结果交付给对方
- 首先是hisat.stat.xlsx这个excel表格里面保存着本次对原始测序数据的比对情况,包括比对率,多比对情况等等。
- 然后是raw_reads_matrix.csv包含着8个样本的近5万个在GENCODE数据库记录着的基因的表达量矩阵。
- 而filter_reads_matrix.csv就是把5万个基因里面过滤掉那些低表达量基因表达量矩阵。
- 最后的rpkm.txt就是把filter_reads_matrix.csv的reads的counts转换为RPKM值,你可以跟paper做比较。
GSM860182_SG.A_correlation.pdf 这个是其中一个比较的结果,可以看出相关性很不错,说明两个流程的结果大致是一致的趋势。
- 如果你进行后续分析的话,可以直接拿rpkm.txt即可。
- 还有那个 mouse_sperm_RNA_seq_bw_files.zip里面是比对的bw文件,可以用IGV打开之后定位具体某个基因看看上面的测序深度的分布情况。
亲爱的粉丝们,大家觉得这个项目,8个样本的RNA-seq数据的分析,值800块钱吗?欢迎留言参与哦!
文末友情宣传
强烈建议你推荐给身边的博士后以及年轻生物学PI,多一点数据认知,让他们的科研上一个台阶:
- 全国巡讲全球听(买一得五) ,你的生物信息学入门课
- 生信技能树的2019年终总结 ,你的生物信息学成长宝藏
- 2020学习主旋律,B站74小时免费教学视频为你领路