第二次出现的错误
踩过坑,才足够刻骨铭心!
这里正好推荐一下这个视频:https://www.bilibili.com/video/av28813815/?p=9
跑完一个RNA-SEQ项目,下意识的看了看bam文件大小,还有最后的文库统计情况,发现非常的诡异,首先是bam文件大小就很奇特:
29M Apr 29 12:15 S12.bam
30M Apr 29 12:15 S1.bam
35M Apr 29 12:10 S22.bam
33M Apr 29 12:10 S2.bam
2.3G Apr 26 18:54 S8.bam
2.2G Apr 26 15:27 S7.bam
2.1G Apr 26 12:05 S4.bam
2.2G Apr 26 06:36 S18.bam
1.8G Apr 26 05:14 S19.bam
2.3G Apr 26 03:35 S6.bam
很明显,4个样本应该是流程失败了,bam文件这么小,最开始推测是不是参考基因组使用错了,但是觉得不应该,这个项目都是同一个物种,不太可能部分样本失败部分样本成功。
然后我看到了文件名很有规律,失败的是 1, 12, 2,22 但是没有一眼看出来为什么,又转向其它项目了,只是把同样的代码重新跑一遍,以为是服务器问题。
因为RNA-SEQ项目我早就搭建好了,很少出这样的幺蛾子,这个坑有点类似于我三年前分享的:
然后是文库统计情况:
| Sample Name | % Assigned | M Assigned |
|---|---|---|
| S14 | 80.30% | 16.1 |
| S15 | 81.10% | 15.8 |
| S20 | 80.00% | 15.4 |
| S13 | 80.10% | 15.2 |
| S6 | 74.70% | 15 |
| S8 | 76.10% | 15 |
| S18 | 77.40% | 14.9 |
| S21 | 80.10% | 14.8 |
| S11 | 77.50% | 14.7 |
| S7 | 79.10% | 14.7 |
| S4 | 79.50% | 14.5 |
| S5 | 78.60% | 14.5 |
| S10 | 76.40% | 13.4 |
| S9 | 78.10% | 13.4 |
| S16 | 76.10% | 12.2 |
| S19 | 79.20% | 12 |
| S3 | 75.50% | 10.6 |
| S22 | 74.40% | 0.3 |
| S1 | 73.70% | 0.2 |
| S12 | 73.60% | 0.2 |
| S2 | 72.90% | 0.2 |
是不是很有趣,看起来这4个样本只是文库偏小而已,并没有其它问题,所以我去检查fq文件,实际情况是他们的fq文件大小相当。
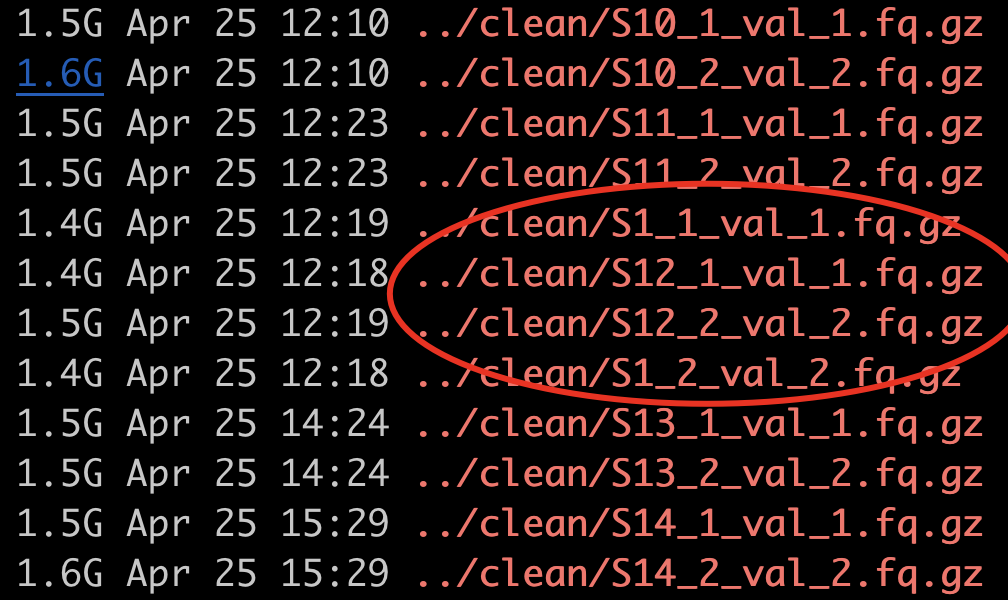
不过我这时候注意到了文件名的排序很诡异,灵光一现,想起来了我录制linux视频的时候,专门提到过的一个问题,就是sort的排序问题。
sort排序问题
这个问题来源于我自己的操作习惯,我制作配置文件一直使用
ls /home/jianmingzeng/rna/raw_data/*1.fq.gz > 1
ls /home/jianmingzeng/rna/raw_data/*2.fq.gz > 2
wc 1 2
cut -d"/" -f 8 1 |cut -d"_" -f 1
cut -d"/" -f 8 1 |cut -d"_" -f 1 > 0
paste 0 1 2 > config
而这个ls的顺序会出现,S1_1.fq.gz 高于 S12_1.fq.gz ,但是呢 S12_2.fq.gz 高于 S1_2.fq.gz 是不是很诡异?
| S1 | S1_1.fq.gz | S12_2.fq.gz |
|---|---|---|
| S12 | S12_1.fq.gz | S1_2.fq.gz |
| S2 | S2_1.fq.gz | S22_2.fq.gz |
| S22 | S22_1.fq.gz | S2_2.fq.gz |
实际上这个问题,我早在linux视频里面讲解过!
关于这个视频为什么大家看不懂
因为要推荐这个系列视频才看了看B站弹幕,发现基本上留言的都是没有看懂的!
不建议这种命名方式
关于实验样本命名,其实我在全国巡讲经常拿出来说事的一个案例就很好的说明了它的重要性。