GEO数据挖掘技巧,基本上该分享的都在B站和GitHub了,目录如下:
- 第一讲:GEO,表达芯片与R
- 第二讲:从GEO下载数据得到表达量矩阵
- 第三讲:对表达量矩阵用GSEA软件做分析
- 第四讲:根据分组信息做差异分析
- 第五讲:对差异基因结果做GO/KEGG超几何分布检验富集分析
- 第六讲:指定基因分组boxplot指定基因list画热图
- 第七讲:根据差异基因list获取string数据库的PPI网络数据
- 第八讲:PPI网络数据用R或者cytoscape画网络图
- 第九讲:网络图的子网络获取
- 第十讲:hug genes如何找
虽然一直演示的表达芯片数据分析,这些芯片分析难点主要是在ID转换,因为不同公司设计的探针命名都不一样,在我4年前博客整理的芯片平台对应R包找:(16)芯片探针与基因的对应关系-生信菜鸟团博客2周年精选文章集
基本上你使用我的标准数据分析代码,下载到表达矩阵,走标准分析流程,火山图,热图,GO/KEGG数据库注释等等,肯定可以出对应的图表。最重要的3张图见:你确定你的差异基因找对了吗?
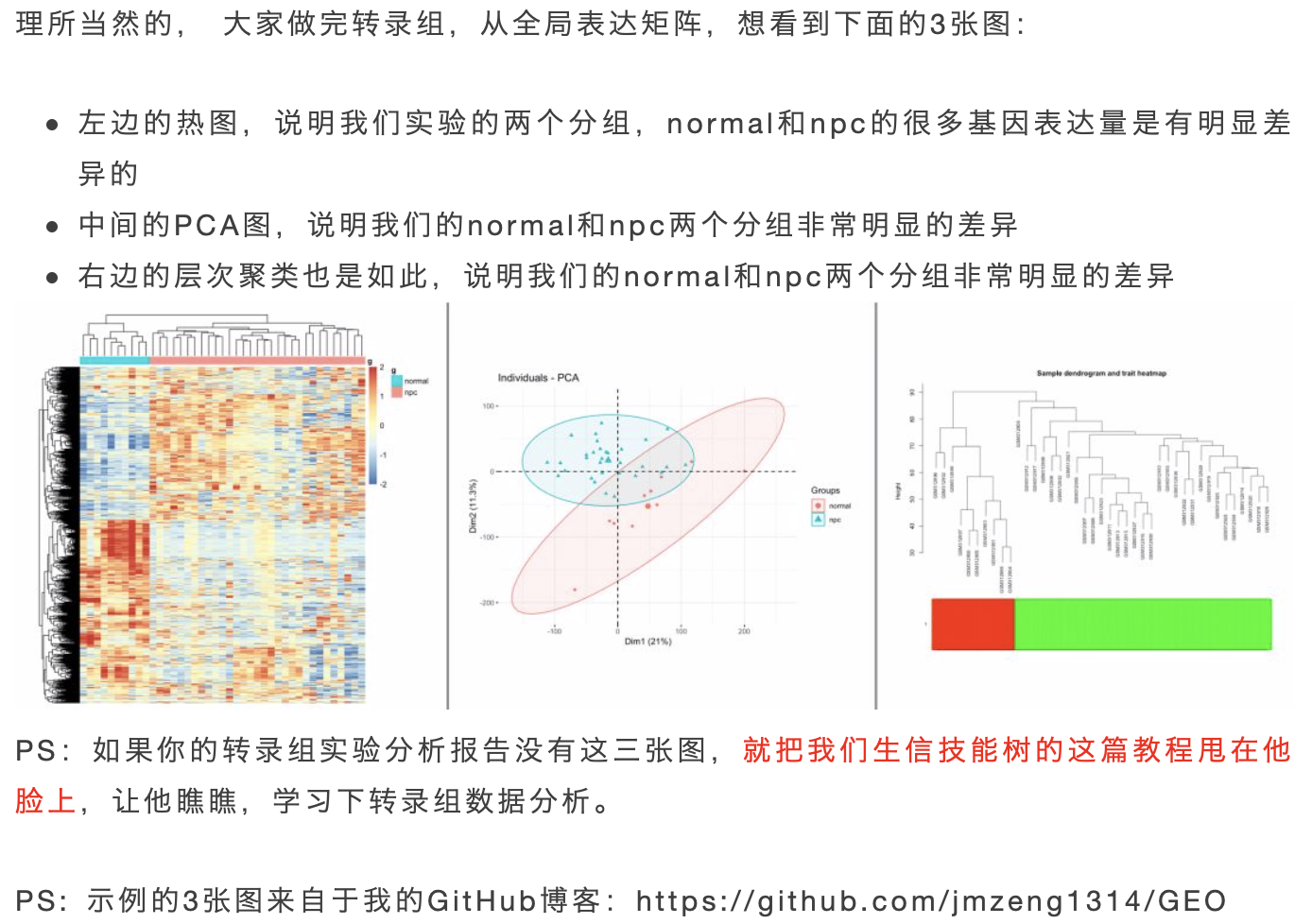
那如果是RNA-seq测序数据呢?
通常呢,RNA-seq测序数据并不会把其表达矩阵存储在Series Matrix File(s) 里面,所以 你使用我的标准代码:
rm(list = ls()) ## 魔幻操作,一键清空~
options(stringsAsFactors = F)#在调用as.data.frame的时,将stringsAsFactors设置为FALSE可以避免character类型自动转化为factor类型
# 注意查看下载文件的大小,检查数据
f='GSE103611_eSet.Rdata'
# https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE103611
library(GEOquery)
# 这个包需要注意两个配置,一般来说自动化的配置是足够的。
#Setting options('download.file.method.GEOquery'='auto')
#Setting options('GEOquery.inmemory.gpl'=FALSE)
if(!file.exists(f)){
gset <- getGEO('GSE103611', destdir=".",
AnnotGPL = F, ## 注释文件
getGPL = F) ## 平台文件
save(gset,file=f) ## 保存到本地
}
load('GSE103611_eSet.Rdata') ## 载入数据
class(gset) #查看数据类型
length(gset) #
class(gset[[1]])
gset
# assayData: 352859 features, 48 samples
只需要把上面的GSE号替换即可,当然如果你不懂GSE号,就需要再细读表达芯片的公共数据库挖掘系列推文感兴趣的也可以去看看;
- 解读GEO数据存放规律及下载,一文就够
- 解读SRA数据库规律一文就够
- 从GEO数据库下载得到表达矩阵 一文就够
- GSEA分析一文就够(单机版+R语言版)
- 根据分组信息做差异分析- 这个一文不够的
- 差异分析得到的结果注释一文就够
比如对 https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE106292 上面的代码就拿不到表达矩阵
因为,这个是RNA-seq数据,作者会把自己的表达矩阵变成Excel表格,方便大家探索!
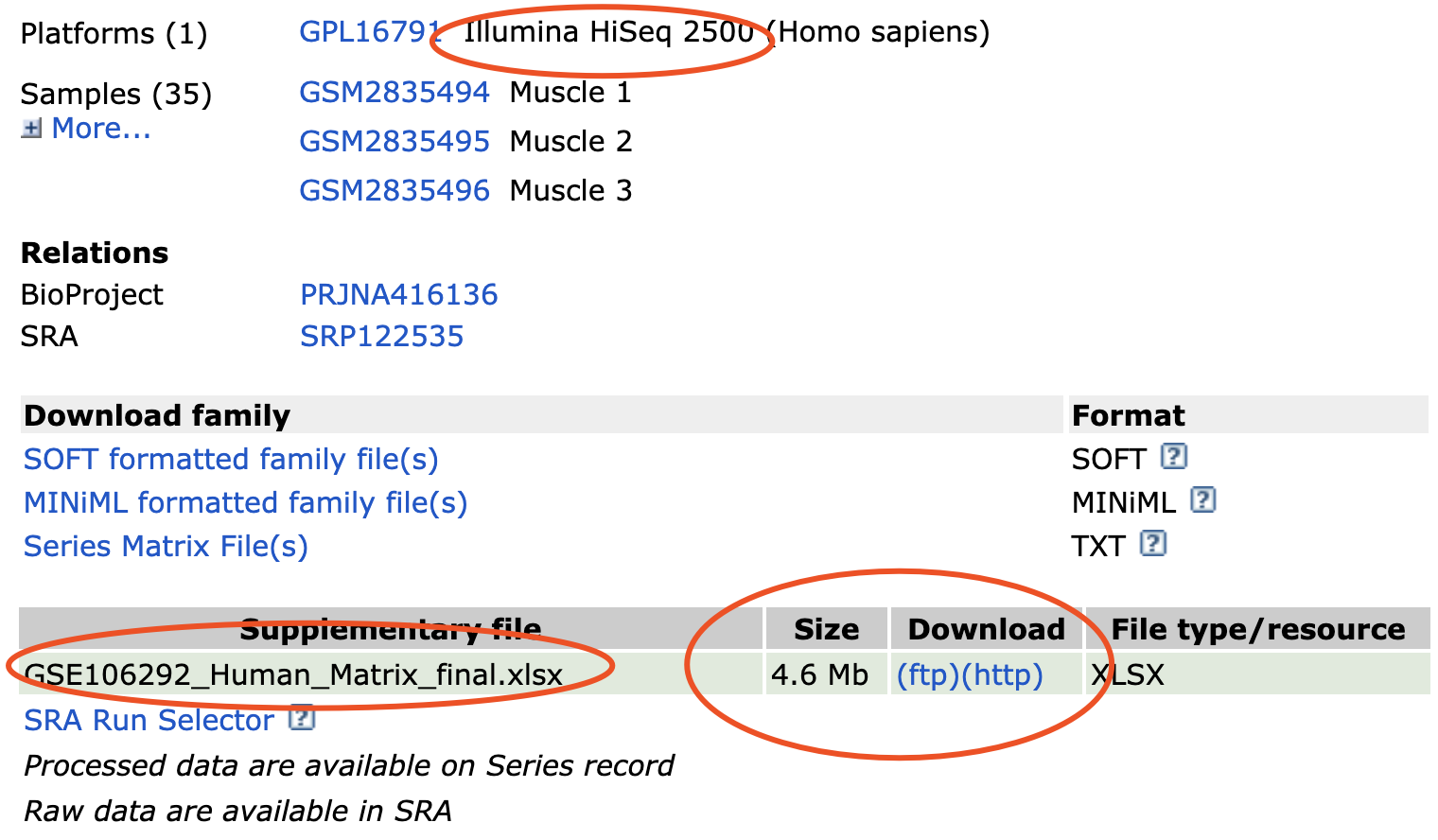
记住,我这里强调了是作者自己的表达矩阵,因为RNA-seq数据分析流程还不一样!参数不一样,软件不一样,数据库不一样,而且最后的表达矩阵的表现形式又不一样!是原始的counts还是RPKM,TPM都不一样!
这里面的知识细节太复杂了,我就不一一展开!建议大家看我们阅读量过10万的RNA-seq系列推文,比如:表达矩阵的归一化和标准化,去除极端值,异常值
现在给大家一个作业
我前面举例的 https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE106292 数据集,其实是一个WGCNA文献,你可以看看下载到的 Excel表格如何读入R里面,做出作者文章的那样的图,可以参考关键问题答疑:WGCNA的输入矩阵到底是什么格式,详细教程见:一文看懂WGCNA 分析(2019更新版)
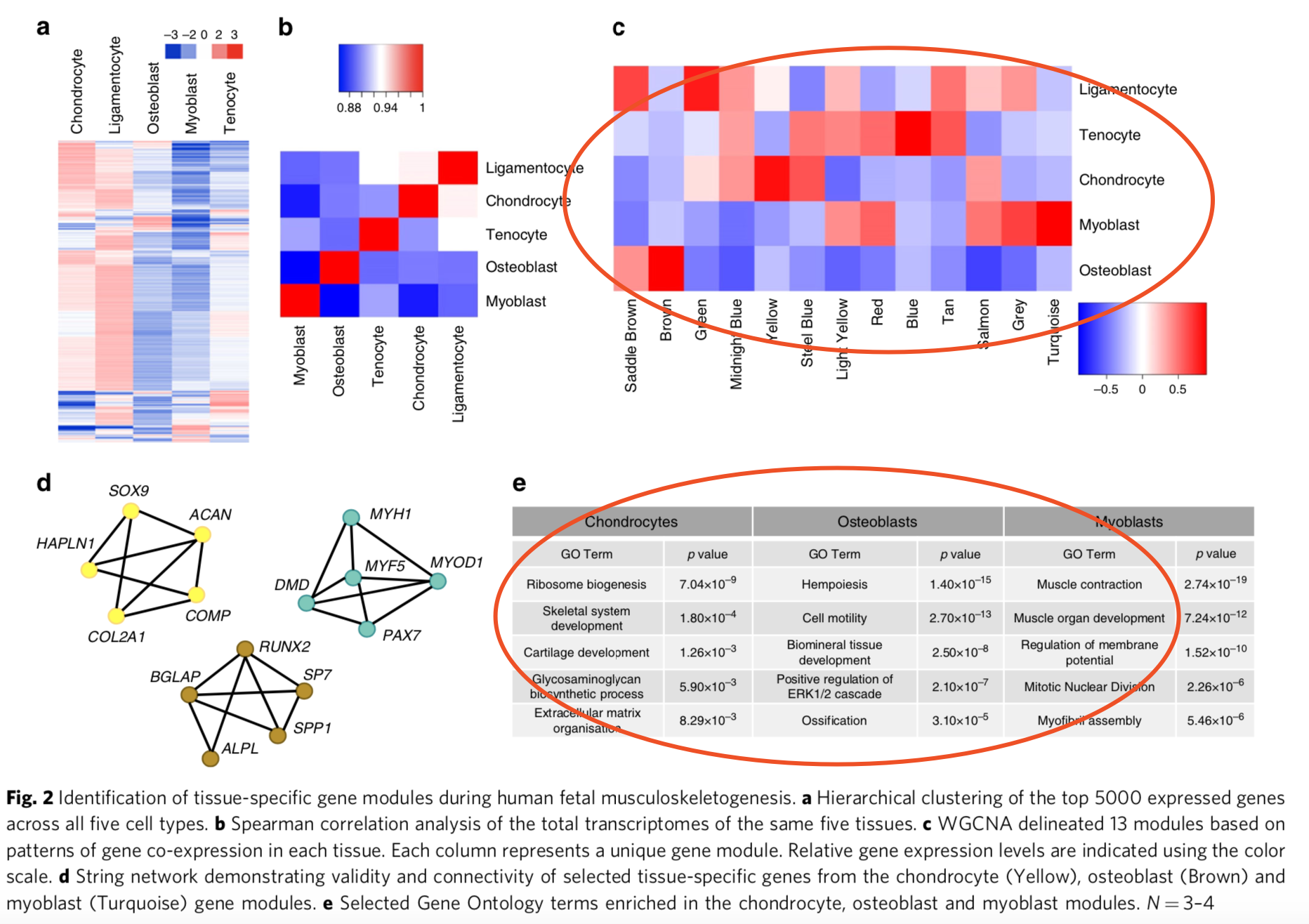
这两个图难度非常大,基本上相当于半年作业的生信工程师经验了,如果你能做出来,发邮件给我你的全部思考分析过程,你可以获得我认可,毕竟相当于有了我7.6%的功力,已经是非常的了不起了!