昨天我们重点强调了star这个比对软件开发团队,附带的star-fusion:最好用的融合基因查找工具终于正式发表了 因为我自己是时隔两年后再次使用它,所以很多数据库和软件代码都没有更新,中间一个小报错就浪费了四五个小时,所以分享一下这个体验!
用过star软件的朋友都知道,参数真的是很多,核心代码是:
start=$(date +%s.%N)
echo star start `date`
$bin_star --runThreadN 4 --genomeDir $star_index \
--twopassMode Basic --outReadsUnmapped None --chimSegmentMin 12 \
--alignIntronMax 100000 --chimSegmentReadGapMax parameter 3 \
--alignSJstitchMismatchNmax 5 -1 5 5 \
--readFilesCommand zcat --readFilesIn $fq1 $fq2 --outFileNamePrefix ${sample}_
mv ${sample}_Aligned.out.sam $sample.sam
$bin_samtools sort -o $sample.bam $sample.sam
$bin_samtools index $sample.bam
$bin_samtools flagstat $sample.bam > $sample.flagstat
touch ok.star.$sample.status
rm $sample.sam
echo star end `date`
dur=$(echo "$(date +%s.%N) - $start" | bc)
printf "Execution time for star : %.6f seconds" $dur
实际上就是一行命令在运行比对过程,但是呢,参数太多了,调起来很麻烦,通常如果不理解的话就不建议修改参数。
学这个软件好些年了,当初把参数弄懂了就一直没有去改变,直到最近需要使用新版star-fusion来找融合基因遇到报错才重新捡起来,报错是:
qiEXITING because of FATAL ERROR: Genome version: 2.7.1a is INCOMPATIBLE with running STAR version: 2.7.0f
SOLUTION: please re-generate genome from scratch with running version of STAR, or with version: 2.7.0d
's
Oct 29 20:10:37 ...... FATAL ERROR, exiting
看起来是版本问题,所以我耗费了约4小时在测试不同的版本,后来发现怎么调整都不对,谷歌搜索看到有一个链接:https://github.com/STAR-Fusion/STAR-Fusion/issues/104 才知道需要注意参数:chimOutJunctionFormat
这个参数默认是0,需要修改为1,大家都知道如果运行软件的时候,使用默认参数就可以不添加,所以我的命令通常是没有修改,那样根本就没有意识到还有这个参数!
比较修改前后软件结果的差异
大家都知道,star软件运行速度很慢,我已经跑了几百个样本,输出了这一点Chimeric.out.junction文件,仅仅是因为一个参数错误,导致其格式并不符合要求,所以我想看看是不是可以比较不同参数的Chimeric.out.junction文件,看看能否修改格式,让它符合star-fusion的输入呢?
首先看,正确的格式:
$head Lib_FUSCCTNBC001_Chimeric.out.junction
chrX 153959404 + chr11 65441502 - -1 0 0 Lib_FUSCCTNBC001.273243 153958692 2S75M564N73M 65441353 149M
chr14 71430437 - chr14 74768794 - 0 0 0 Lib_FUSCCTNBC001.273359 71430438 73S77M 74768544 150M27p73M77S
chr21 8251540 - chr8 42032872 + -1 0 0 Lib_FUSCCTNBC001.273411 8251541 5S145M 42032873 83M
chr2 207113838 + chr2 207113716 + 0 0 5 Lib_FUSCCTNBC001.273783 207113809 29M121S 207113717 29S121M-67p150M
chr19 23653819 + chr19 23653669 + -1 0 0 Lib_FUSCCTNBC001.273973 23653672 147M 23653670 149M
chr5 109336411 + chr14 23312911 - -1 0 0 Lib_FUSCCTNBC001.274024 109336261 150M 23312848 63M
chr6 7289920 + chr2 215980809 + 0 0 0 Lib_FUSCCTNBC001.274048 7289823 97M53S 215980810 97S53M-14p74M
chr11 33287179 - chr13 19794055 + 0 0 0 Lib_FUSCCTNBC001.274095 33287180 35S115M 19794056 115S35M-35p145M
chr13 60042810 + chr12 56597247 + -1 0 0 Lib_FUSCCTNBC001.274103 60016117 1S29M26544N120M 56597248 137M
chr8 22649163 + chr8 22648954 + 0 0 7 Lib_FUSCCTNBC001.274137 22649109 54M96S 22648955 54S96M57p91M
然后看错误的格式:
$head SRR2016957_Chimeric.out.junction
chr17 58331130 - chr17 58331230 - -1 0 0 SRR2016957.400942 58331131 3S97M 58331131 99M1S
chr1 201331532 - chr17 1033652 + -1 0 0 SRR2016957.400994 201331533 100M 1033653 100M
chr20 41414170 - chr20 41414134 + 0 0 2 SRR2016957.401127 41414171 33S67M-40p100M 41414135 67S33M
chr14 102810820 + chr14 102811010 - -1 0 0 SRR2016957.401161 102810720 100M 102810913 97M3S
chr13 95612067 - chr13 95619886 + 0 0 2 SRR2016957.401169 95612068 25S75M7777p28M2395N72M 95619887 75S25M
chr1 3889730 + chr10 62087322 + 0 0 1 SRR2016957.401243 3889419 100M183p28M25S 62087323 28S25M
chr7 33155691 + chr7 33257368 - 0 0 1 SRR2016957.401278 33155644 47M53S 33177481 100M79734p53M47S
chr17 7513826 - chr17 7513925 - -1 0 0 SRR2016957.401300 7513827 1S97M 7513827 98M
chr8 100272833 - chr17 28602758 - -1 0 0 SRR2016957.401332 100272834 100M 28602659 99M
chr2 233516919 - chr2 233516874 + 0 0 4 SRR2016957.401361 233516920 44S56M160p100M 233516875 56S44M
是不是傻眼了,居然一模一样的格式,那我修改这个参数干啥???
仔细看说明书文档
调用命令查看说明书文档,发现:
chimOutJunctionFormat 0
int: formatting type for the Chimeric.out.junction file
0 ... no comment lines/headers
1 ... comment lines at the end of the file: command line and Nreads: total, unique, multi
原来这个命令,仅仅是在文件末尾加上两个井号键开头的注释信息,说不定star-fusion软件本来就不使用这个信息呢,仅仅是看看文末有没有两个井号键开头的注释信息来判断我们的star软件是否合格!
那我们现在看看这个参数修改后的Chimeric.out.junction文件增加的两个井号键开头的注释信息到底是什么吧:
# 2.7.3a /home/yb77613/biosoft/STAR-2.7.3a/bin/Linux_x86_64/STAR --runThreadN 4 --genomeDir /home/yb77613/biosoft/starFusion/db/GRCh38_gencode_v31_CTAT_lib_Oct012019.plug-n-play/ctat_genome_lib_build_dir/ref_genome.fa.star.idx/ --twopassMode Basic --outReadsUnmapped None --chimSegmentMin 12 --alignIntronMax 100000 --chimSegmentReadGapMax parameter 3 --chimOutJunctionFormat 1 --alignSJstitchMismatchNmax 5 -1 5 5 --readFilesCommand zcat --readFilesIn /home/yb77613/data/public/tnbc/clean/Lib_FUSCCTNBC001_1_val_1.fq.gz /home/yb77613/data/public/tnbc/clean/Lib_FUSCCTNBC001_2_val_2.fq.gz --outFileNamePrefix Lib_FUSCCTNBC001_
# Nreads 41047586 NreadsUnique 31992503 NreadsMulti 6000335
可以看到,第一个跟bam文件的头注释信息差不多,就是记录着我们的软件命令,但是第二行稍微有点不一样,信息,而且每个样本的信息都不一样:
Lib_FUSCCTNBC001_Chimeric.out.junction:# Nreads 41047586 NreadsUnique 31992503 NreadsMulti 6000335
Lib_FUSCCTNBC003_Chimeric.out.junction:# Nreads 22899117 NreadsUnique 19079724 NreadsMulti 2349946
Lib_FUSCCTNBC003.PT_Chimeric.out.junction:# Nreads 34291126 NreadsUnique 27037183 NreadsMulti 4164969
Lib_FUSCCTNBC004_Chimeric.out.junction:# Nreads 32166763 NreadsUnique 21336913 NreadsMulti 7887447
两个策略
首先,我们可以去看star-fusion软件读取Chimeric.out.junction文件的perl代码,看看软件到底需要这文件末尾加上两个井号键开头的注释信息干嘛了,其次,我们可以尝试修改这文件末尾加上两个井号键开头的注释信息,看看运行star-fusion软件是否结果出现不一样的地方。
查看源代码毕竟难道较高,我们先走第二个策略,尝试修改这文件末尾加上两个井号键开头的注释信息,看看修改前后结果是否有差异
STAR-Fusion --genome_lib_dir $lib -J Lib_FUSCCTNBC001_Chimeric.out.junction --output_dir s1
我简单看了看,实际上并没有差异, 但是比较结果的差异其实也是件很复杂的事情,实际上查源代码是最肯定的解决方案,如果公司有这样的人物是最好的啦!
如果两个策略的工程师公司或者科研团队都没有,还有一条路,就是全部推倒重来,只要你的计算资源足够,时间也足够,无非就最多一个星期而已!
关于软件设计的一点看法
多说一句,我特别不喜欢软件在输出里面加上这些小尾巴,比如htseq-counts,在TCGA下载表达矩阵,就会出现:
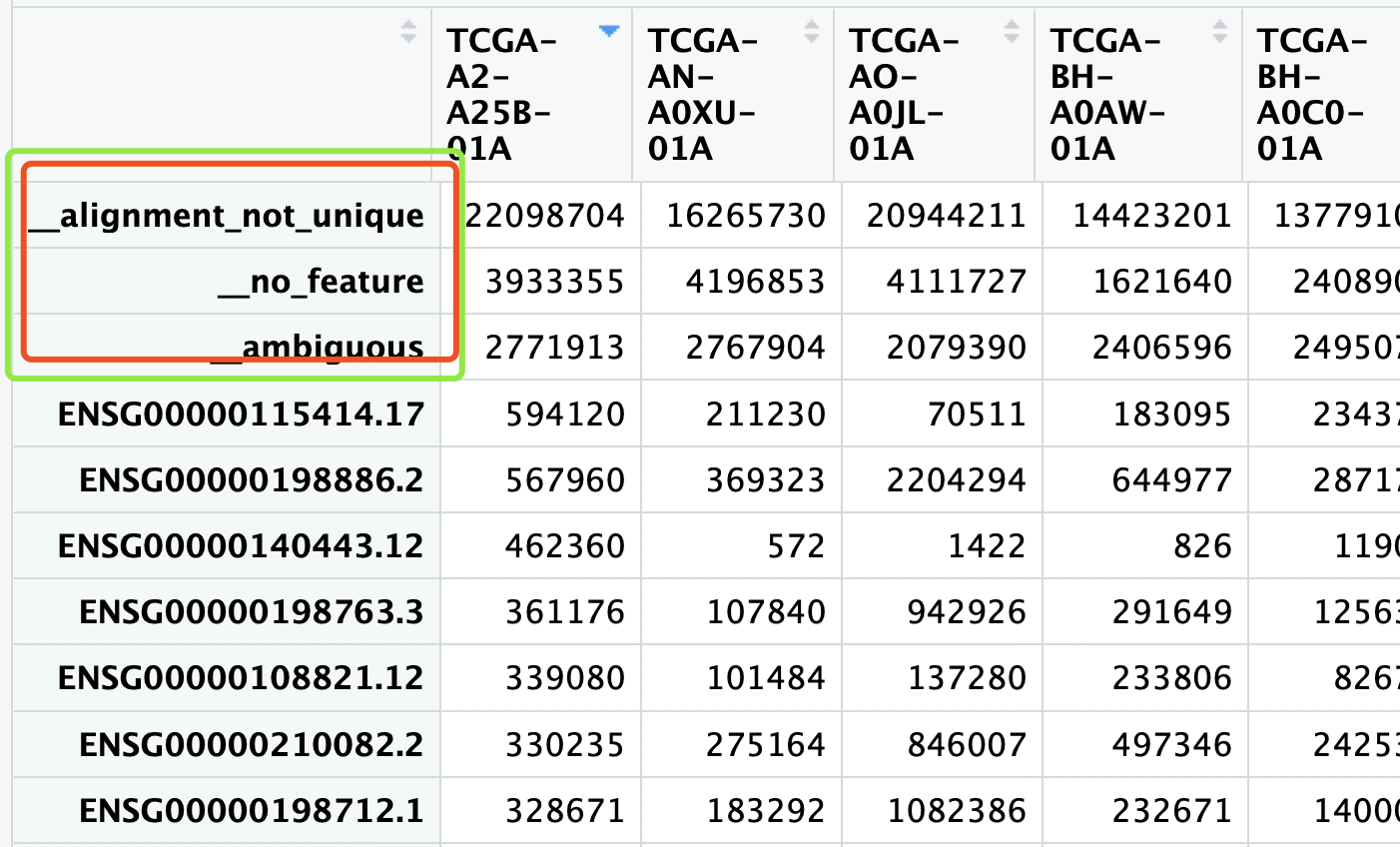
一般人技术不行,也不够细心,根本就无法意识到,里面有在这些不是基因的玩意!
另外关于star-fusion软件的一个提议
大家都知道,目前单细胞是10x的天下,而10x的测序数据,御用软件cellranger其实就是star的包装,关于10X仪器的单细胞转录组数据走cellranger流程,我们在单细胞天地多次分享过流程笔记,大家可以自行前往学习,如下:
- 单细胞实战(一)数据下载
- 单细胞实战(二) cell ranger使用前注意事项
- 单细胞实战(三) Cell Ranger使用初探
- 单细胞实战(四) Cell Ranger流程概览
- 单细胞实战(五) 理解cellranger count的结果
我这里想说的是,既然是star的包装,其实结果就可以走star-fusion来找融合基因,好奇怪的是目前大量的单细胞转录组数据出来了,却没有一个文章去探索融合基因,也没有人开发工具,是一个空白市场,大家可以试试看哦。
不过,商业化很成功的10X仪器做单细胞其实找融合基因还是有点勉强的,毕竟它并不是转录组全长测序,所以基本上很难获得融合位点融合事件,不过,如果是smart-seq2技术实际上是可以的啊!。不要仅仅是走单细胞下游分析标准流程啊,就是那些R包的认知,包括 scater,monocle,Seurat,scran,M3Drop 需要熟练掌握它们的对象,:一些单细胞转录组R包的对象 ,分析流程也大同小异:
- step1: 创建对象
- step2: 质量控制
- step3: 表达量的标准化和归一化
- step4: 去除干扰因素(多个样本整合)
- step5: 判断重要的基因
- step6: 多种降维算法
- step7: 可视化降维结果
- step8: 多种聚类算法
- step9: 聚类后找每个细胞亚群的标志基因
- step10: 继续分类
你可以做的更好!