最近收到网友求助,问:
尝试一篇文献的表达差异分析和热图重现,主要参考您Github中GEO-master/GSE42872_main的代码,但我跑出的差异分析列表logFC与文献给出的列表数据不符,尝试了很多次,不清楚是什么原因?
本来我一般是不理会这样的求助的, 毕竟代码都给了,还不会用,总不能怪我了,冥冥中鬼使神差的回复了:
问题在哪里,我就没得空去帮你检查,你要是真想我回答,两个办法。
第一个是把你这篇文献写一个PPT,介绍这方面背景知识点给我,我学习到了新知识,作为交换,我就帮你修改代码
第二个是,你直接付费我来帮你检查代码
有趣的是,对方马上甩来了一个详细的PPT,让我也学到了知识,所以就投桃报李,帮忙检查代码,结果发现很有趣的事情,就是这个数据集的作者,居然上传了错误的表达矩阵。
错误的表达矩阵

[HG-U133_Plus_2] Affymetrix Human Genome U133 Plus 2.0 Array 这个芯片平台怎么可能只有不到五千个探针!
所以肯定是作者上传错了,因为文件大小就不对。

下载CEL文件
这个时候必须要下载原始数据了。
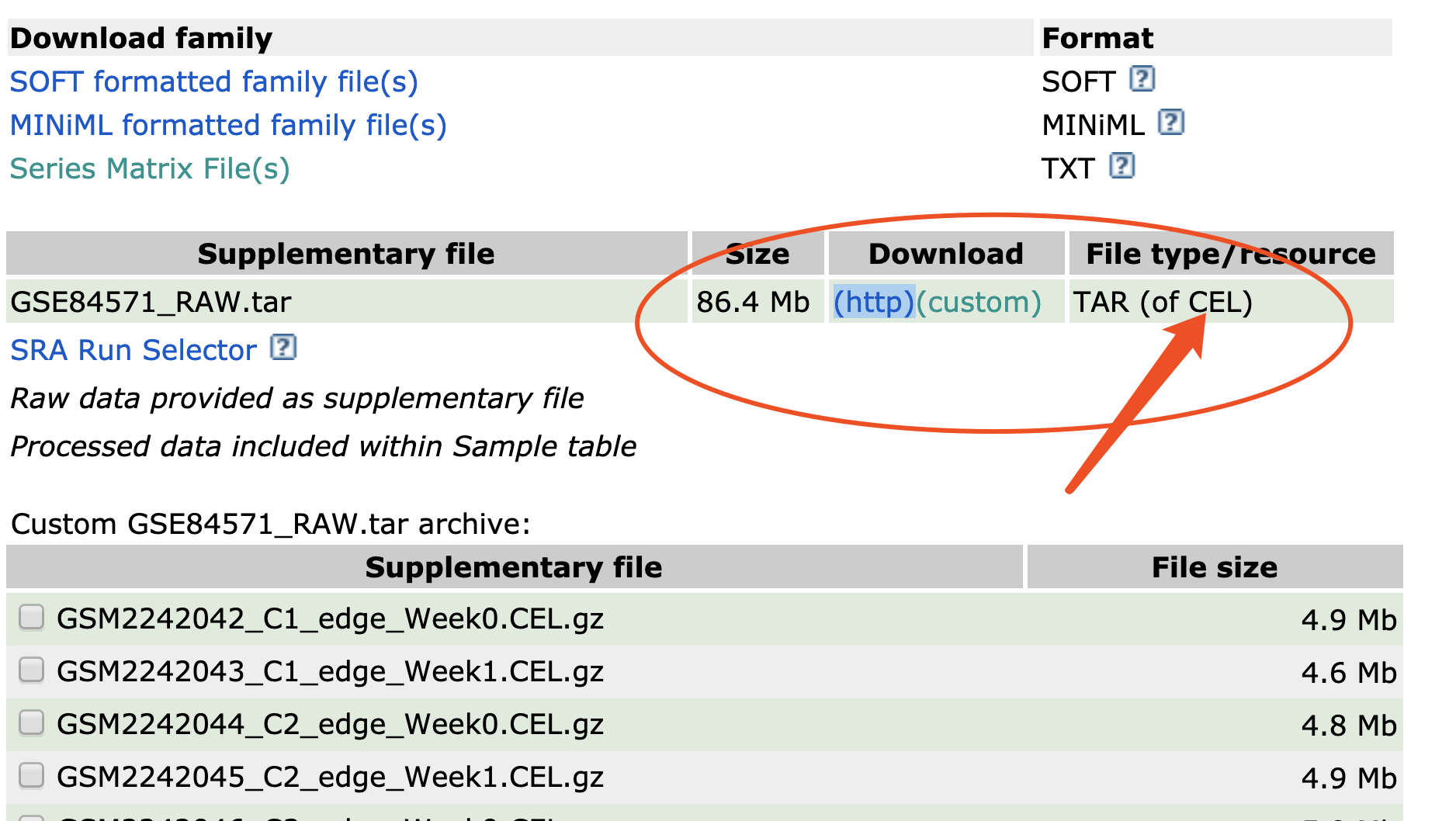
拿到芯片的原始数据,cel文件的下载地址:ftp://ftp.ncbi.nlm.nih.gov/geo/series/GSE84nnn/GSE84571/suppl/GSE84571_RAW.tar
下载这个文件很坎坷,还是我们伟大的墙,解压后就处理。
处理CEL文件
这个是我翻开三年前自己整理的代码,参考我的GEO教程,在https://github.com/jmzeng1314/GEO 上面
# BiocManager::install(c( 'oligo' ),ask = F,update = F)
library(oligo)
# BiocManager::install(c( 'pd.hg.u133.plus.2' ),ask = F,update = F)
library(pd.hg.u133.plus.2)
dir='~/Downloads/GSE84571_RAW/'
od=getwd()
setwd(dir)
celFiles <- list.celfiles(listGzipped = T)
celFiles
affyRaw <- read.celfiles( celFiles )
setwd(od)
eset <- rma(affyRaw)
eset
# http://math.usu.edu/jrstevens/stat5570/1.4.Preprocess_4up.pdf
save(eset,celFiles,file = f)
# write.exprs(eset,file="data.txt")
得到的eset这个对象,与我们之前一直讲解的GEOquery包下载是一样的, 所以后续代码不需要变化。
得到表达矩阵和表型信息
a=eset
dat=exprs(a) #a现在是一个对象,取a这个对象通过看说明书知道要用exprs这个函数
dim(dat)#看一下dat这个矩阵的维度
# [HG-U133_Plus_2] Affymetrix Human Genome U133 Plus 2.0 Array
dat[1:4,1:4] #查看dat这个矩阵的1至4行和1至4列,逗号前为行,逗号后为列
#dat=log(dat)
dat[1:4,1:4]
boxplot(dat[,1:4])
colnames(exprs)
group_list = paste(substring(celFiles,12,12),
substring(celFiles,24,24),sep = '-')
table(group_list) #统计频率
save(dat,group_list,file = 'step1-output.Rdata')
有了表达矩阵和表型信息后续分析就完全参考我https://github.com/jmzeng1314/GEO代码即可。
差异分析结果跟作者的附件公布的数量相当,基因也基本一致,问题就解决啦。