写在前面
你现在看到的是文献俱乐部2019年笔记分享第一弹,我将会在春节7天连续分享,目录如下:
2019年1月份第1周(总第49周)单细胞转录组探索CAFs的功能和空间异质性
2019年1月份第2周(总第50周)异常CRC病人的突变时空异质性与免疫
2019年1月份第4周(总第52周)TCGA计划的ATAC-seq数据发布
2019年2月份第2周(总第54周)测173个成年人的大脑的102个基因
2019年2月份第3周(总第55周)2.5万汉族人的GWAS乳腺癌风险基因
因为学业需要,我阅读的大量文献都是NGS组学相关,所以会涉及到很多数据处理,而这些文献基于的生物信息学数据处理技巧,我都在过去的5年里以各种形式分享讲解过,也有系列视频,希望你可以在方便的时候再次学习一遍,查漏补缺。也欢迎推荐给有需要的朋友
学习笔记目录
1.3个学生的linux视频学习笔记
2.生信人应该这样学R语言系列视频学习心得笔记分享
3.一万人陪你学习GEO数据库挖掘知识(公益视频听课笔4.记分享)
5.公共数据库挖掘视频学习心得体会
6.生信小技巧系列第一季完结版视频教程学习笔记分享
……期待有你……
如果,你不仅仅是对NGS组学应用文献感兴趣,也欢迎加入我们文献阅读小组分享自己的主页领域文献。
逆向收费读文献社群(第二年通知)
今天是大年初七,给大家带来的是2.5万汉族人的GWAS乳腺癌风险基因,希望你能学到知识。
2.5万汉族人的GWAS乳腺癌风险基因
文章发表于June 2018 在著名肿瘤学期刊 Cancer Research ,题目是:A Large-Scale, Exome-Wide Association Study of Han Chinese Women Identifies Three Novel Loci Predisposing to Breast Cancer 尽管全基因组关联研究(GWAS)已经确定了超过90个乳腺癌易感位点,仍然存在显而易见的遗传力丢失,并且尚未系统地评估编码区突变对乳腺癌易感性的贡献。
安徽医科大学第二医院肿瘤科以及上海复旦大学上海癌症中心等机构合作开展的由24,162个个体(10,055例病例和14,107个对照)组成的大规模全基因组乳腺癌关联研究。已知的易感性位点(例如ESR1,FGFR2和TOX3)在此次研究中得到了重复。
同时还 鉴定出了两个全新的编码变体:C21orf58和ZFN526,以及一个新的位于7q21.11的非编码变体,它们在控制乳腺癌细胞生长中具有功能性作用。 该研究揭示了乳腺癌遗传易感性的新基因和位点,拓展了乳腺癌的复杂基因遗传学背景;同时在临床上,扩大了肿瘤基因筛查,尤其是针对汉族女性乳腺癌基因筛查的潜在基因选择范围。
外显子芯片做GWAS
这里研究团队选择的是定制化的 Illumina Human Exome Asian BeadChip (Exome_Asian Array). 还有网页关于此芯片的介绍:(http://genome.sph.umich.edu/wiki/Exome_Chip_Design).
课题设计
尽管已经有了不少乳腺癌GWAS大队列研究发表,本研究的样本量仍然算是很可观了。
不知道这些芯片数据在哪里,如果能收集到受试者更多信息,这几万人的芯片数据可以作为一个商业消费者基因检测公司的起家数据资本了。
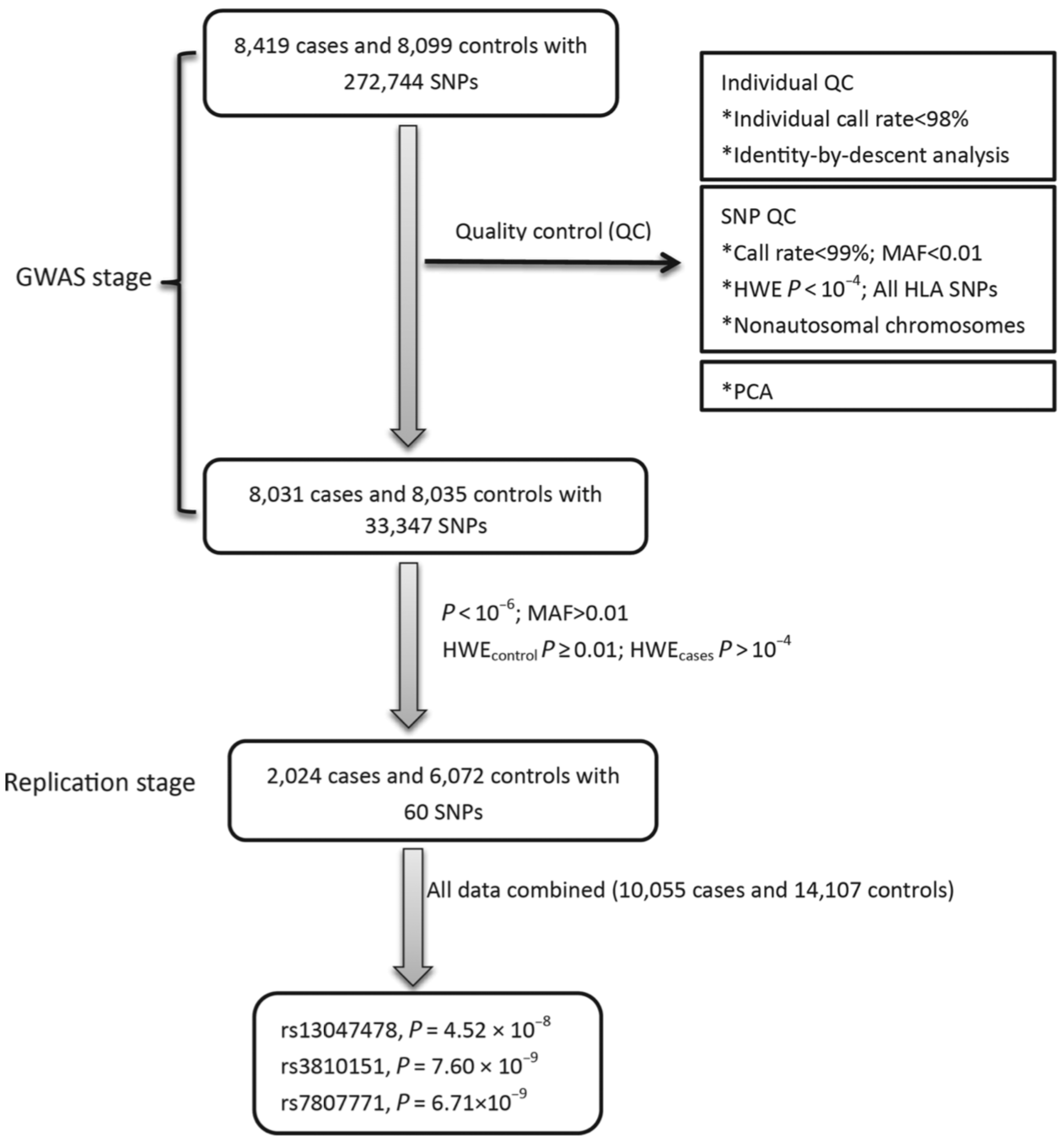
芯片数据分析流程
首先是质控
- 样本的过滤
- 位点的过滤
这些质控标准,上面的流程图有列出部分,这个是GWAS数据分析部分的精华,合理的质控。
还使用了Sequenom MassARRAY system (Sequenom, Inc.) and Multiplex SnapShot technology (Applied Biosystems, Inc.对挑选到的60个位点进行验证。曼哈顿图确定GWAS显著位点
曼哈顿图就是使用二维散点图展示大量数值的办法。最普遍的应用就是在全基因组关联分析(GWAS)中,展示全基因组水平所有SNP的与某个性状相关性的P value。
R包“qqman”就可以做manhattan图。
曼哈顿图的X轴是对应的SNP在染色体上的坐标,从图中颜色就可以看出坐标是按照染色排序的。
对应的Y轴是SNP的P value的-log10值。例如,P value=0.001,对应的-log10(p value)=3。所以,Y轴值越大,说明对应SNP的P value越显著。图中的红色横线就是SNP 显著性的阈值线,超过的这条阈值的位点就是显著的关联性位点。
在本研究就,超过阈值的就3个SNP位点,是两个全新的编码变体:C21orf58和ZFN526,以及一个新的位于7q21.11的非编码变体
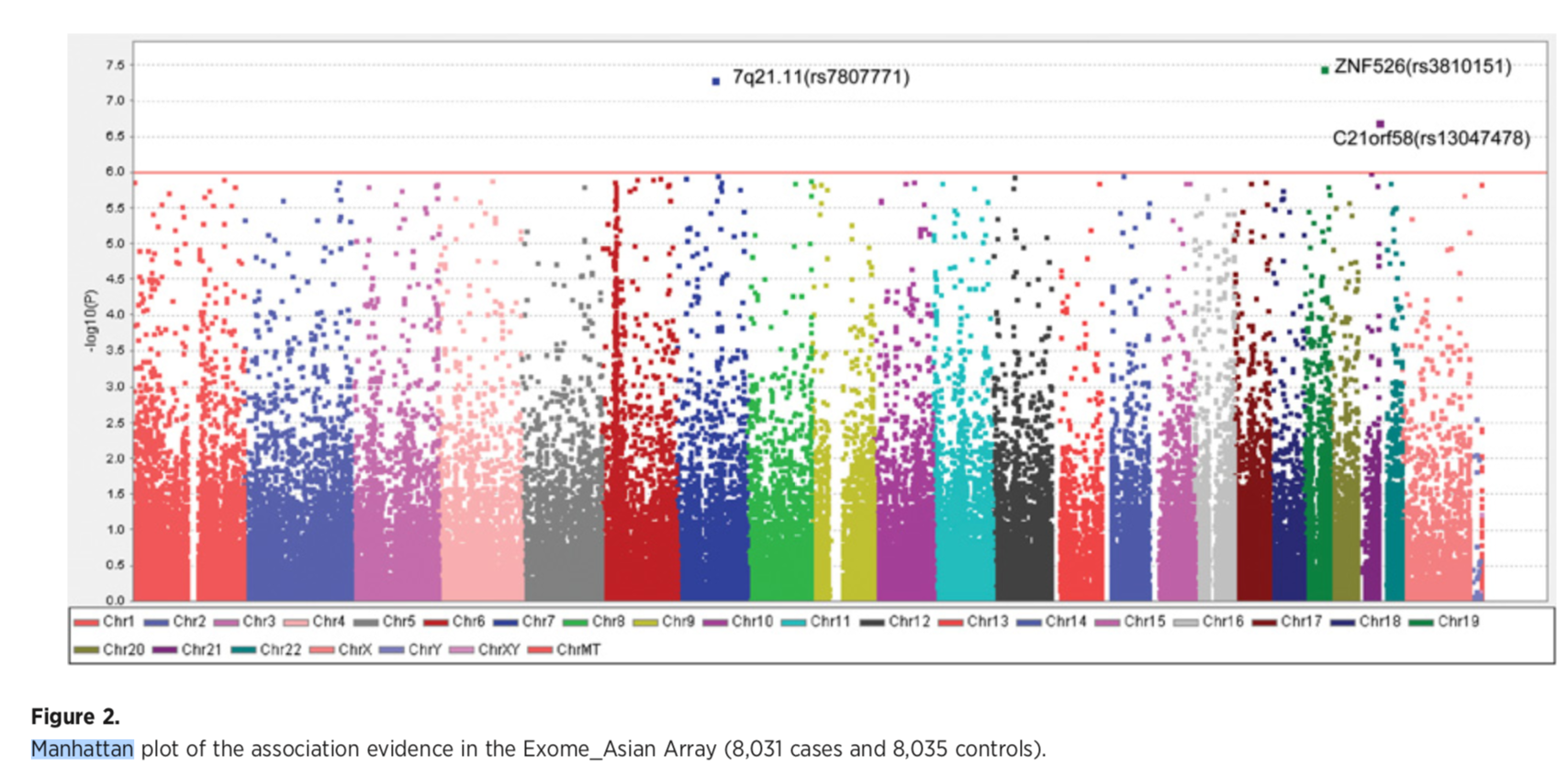
说明GWAS找到的性状相关位点的功能
GWAS定位到具体的SNP位点是生物学故事的精彩之处,但为了让故事更加丰满,继续探索,实验验证等下游分析也是必不可少的。
作者的GWAS研究,定位到了 两个全新的编码变体:C21orf58和ZFN526,以及一个新的位于7q21.11的非编码变体 。 - rs13047478-associated gene C21orf58 and the rs3810151-associated ZNF526
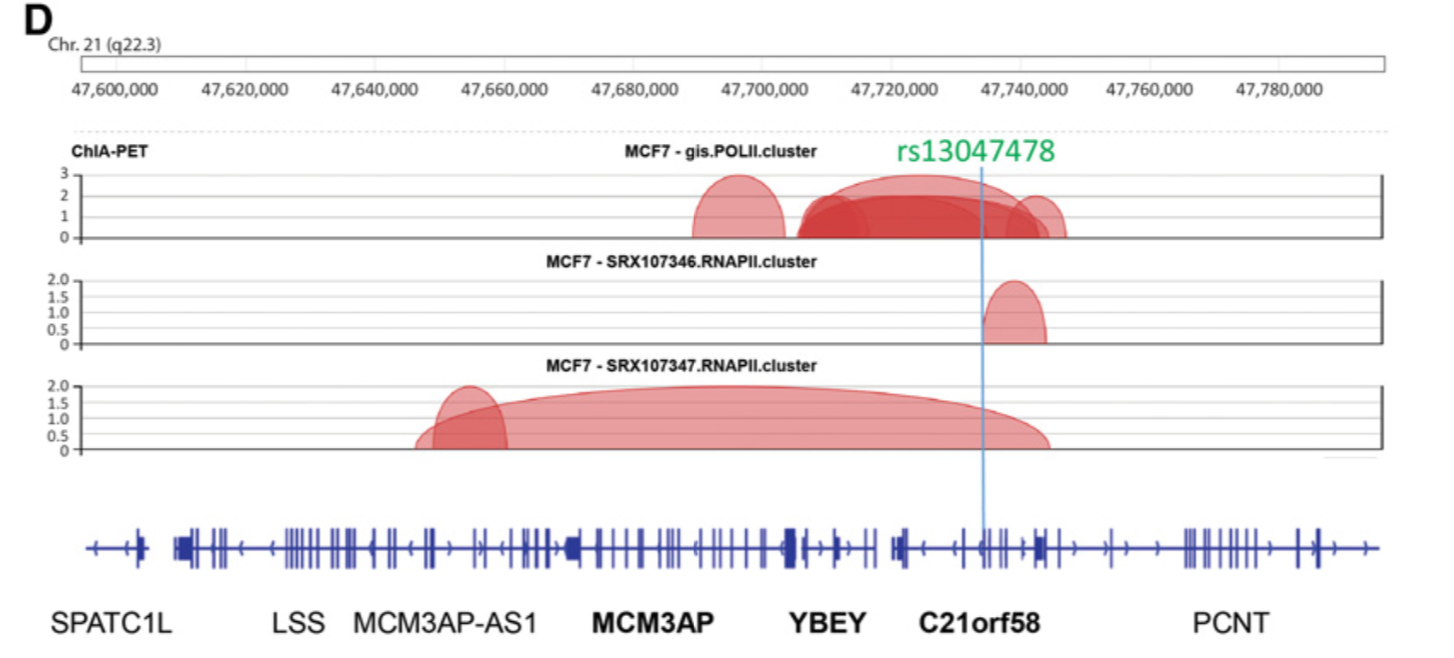
使用 Luciferase reporter assays 实验,可以说明在rs13047478位点,对MCM3AP来说,A碱基相比G碱基来说,增加表达。而YBEY相反。
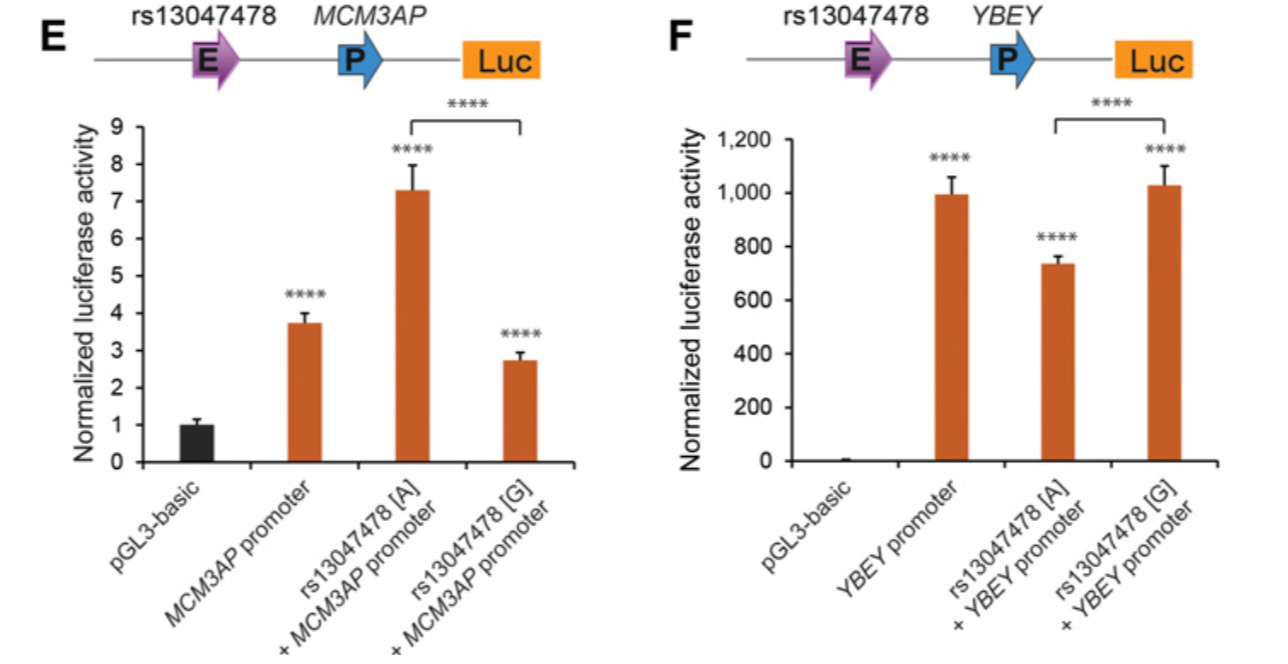
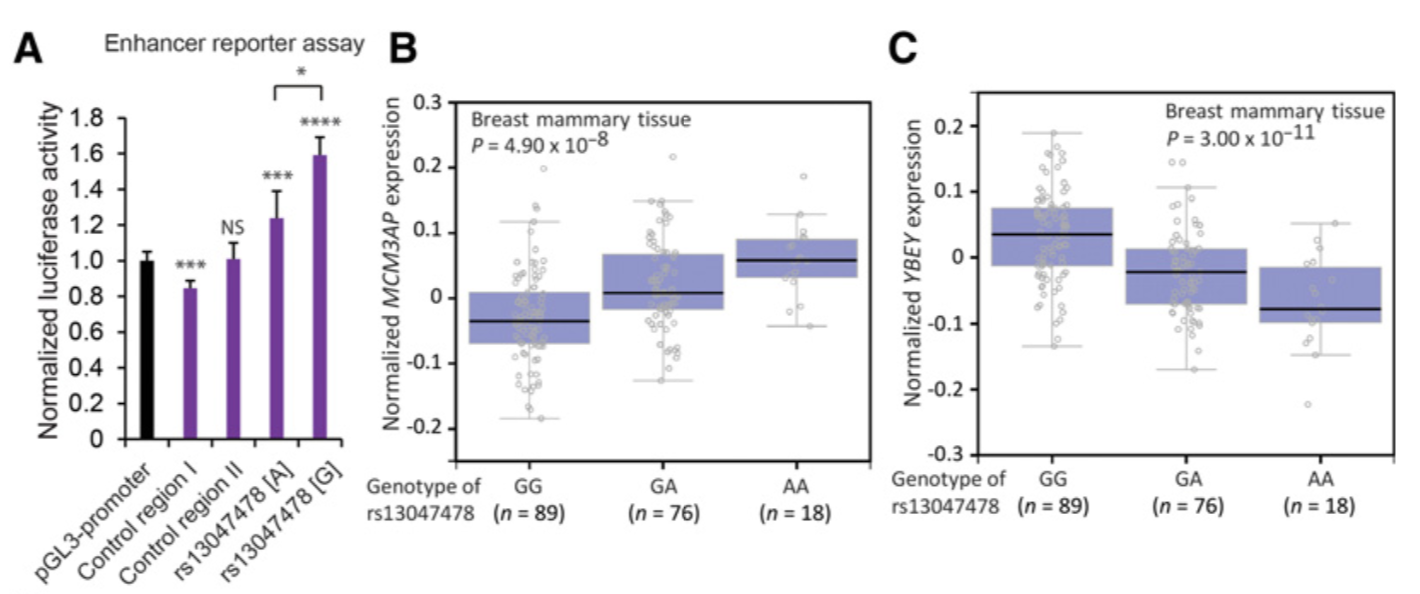
同样是Luciferase reporter assays 说明rs13047478位点的两个碱基增强子能力都表现了,高于随机区域碱基,也高于 pGL3-promoter 空载的增强表达能力。而且GTEX数据库说明rs13047478位点的不同碱基形式与MCM3AP和YBEY基因表达是显著相关的。
已知 BRD4, CCND1, and MYC 基因对乳腺癌细胞生长是必须的,而rs13047478-associated gene C21orf58 and the rs3810151-associated ZNF526 也是乳腺癌细胞生长依赖的。
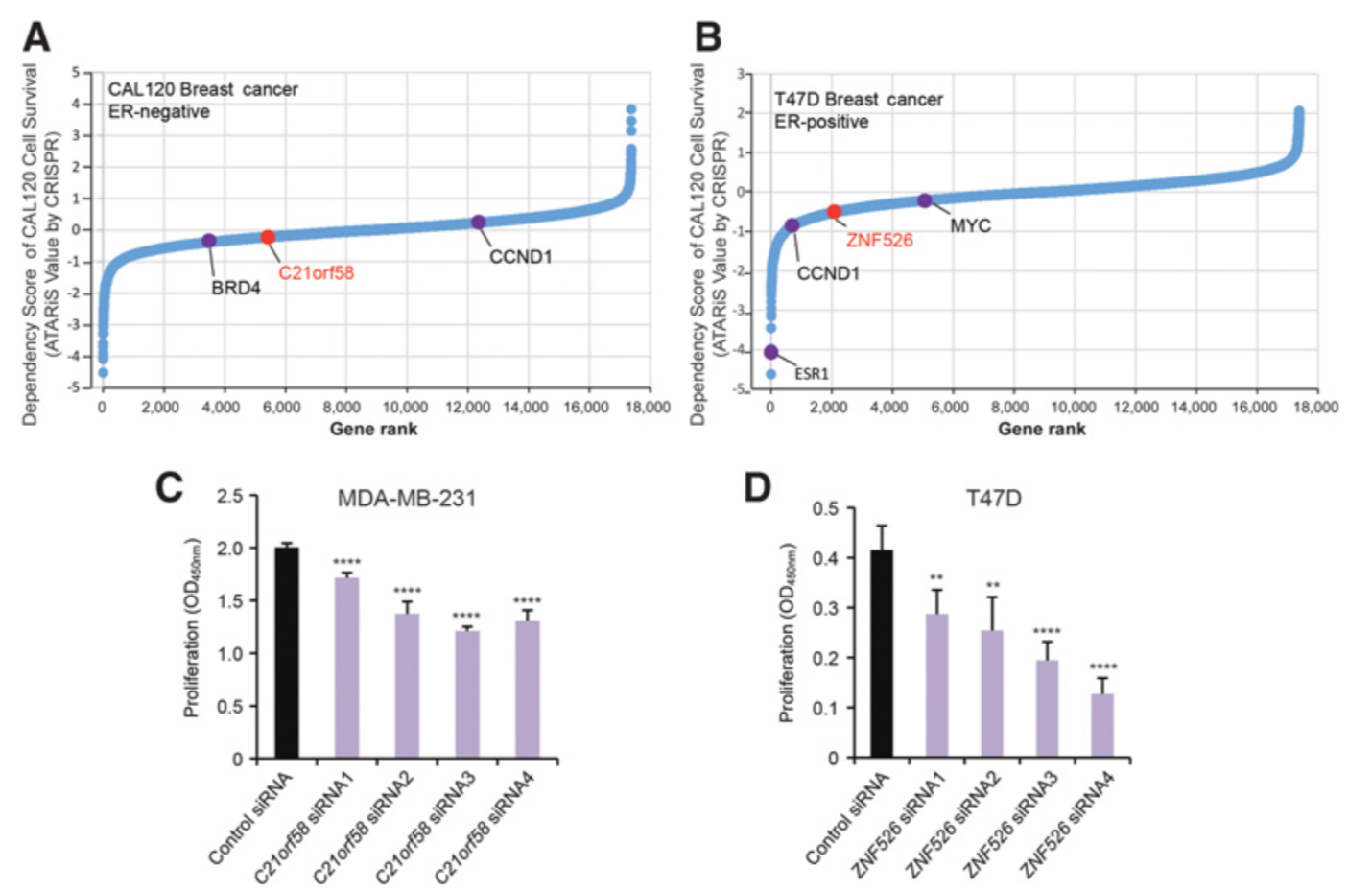
公共数据库检测表达量
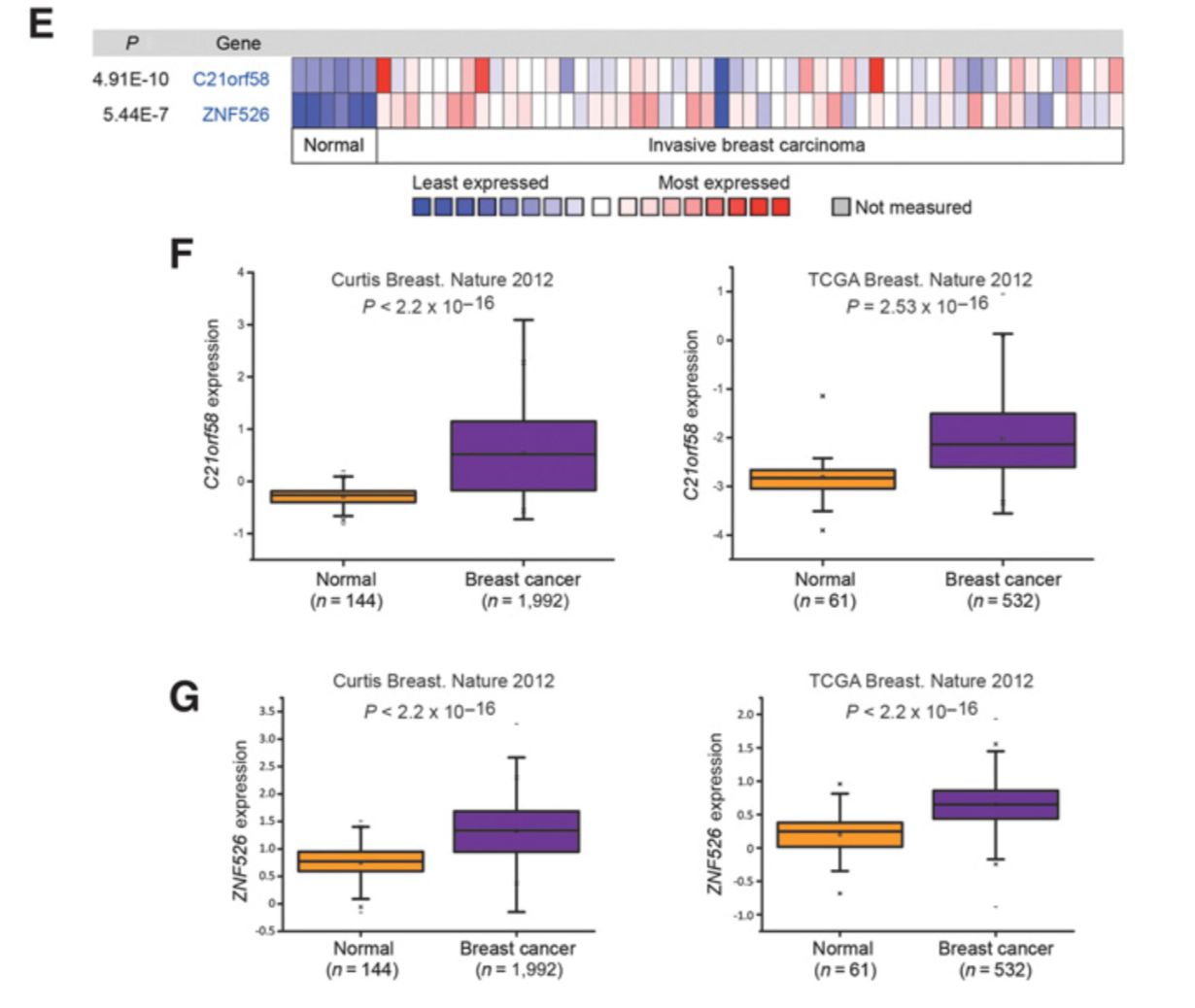
同样是 对 rs13047478-associated gene C21orf58 and the rs3810151-associated ZNF526 在TCGA等各种乳腺癌相关数据库看表达量。
可以看到,这两个基因的表达量在癌症病人都是显著高于正常组织的,不仅仅是在TCGA数据集,在其它已经发表的大文章人群队列也是如此。
三维基因组结构
这里使用了ChIA-PET公共数据看指定变异位点。
乳腺癌的GWAS历史
GWAS(Genome-wide association study,全基因组关联分析)的设想由Neil Risch在1996年提出。GWAS在全基因组范围内比较患者和对照组的SNP(Single Nucleotide Polymorphism)位点基因型性的分布,找出所有的变异等位基因频率,从而避免了像候选基因策略一样需要预先假设致病基因。
在应用GWAS这个分析方法时有几个关键点需要把握: - 一是数据预处理,比如需要对基因芯片产生的数据进行仔细处理以消除人为因素造成的差异;
- 二是一些表型在人种之间差别较大,需要区分研究;
- 三是卡方检验要求的p值阈值很小,比如为5乘以10的-8次方,也就是说1亿次实验只允许有5次犯错,这就对信号的要求非常高;
- 四是不同的基因之间可能会有很强的关联性,这时候需要结合生物学信息和合适的统计学方法对这些关联加以分析和鉴别。
因为GWAS只分析多态性位点(MAF>5%),那么通过分析低频位点(0.5%<MAF<5%)与罕见位点(MAF<0.5%)也许能够解释更多的遗传因素,罕见位点已知与人类很多疾病相关,比如单基因遗传病或常见疾病的罕见种类(如遗传性乳腺癌),从进化角度来说这些罕见位点导致的遗传病将会被筛选掉,但是最近有一些研究已经发现低频位点与罕见位点在常见疾病发病过程中发挥重要作用。
GWAS 发现的复杂疾病相关的遗传变异,即单核苷酸多态性(SNP)位点大多位于基因的非编码区,并且同一区域中连锁的遗传变异(SNP)位点可以多达成百上千个,如何从中找到真正与疾病相关的SNP,并从生物学上诠释其功能及其与疾病的关系,是后GWAS 时代的重大挑战之一。2015年的3月9日- meta分析
剑桥大学遗传流行病学教授Doug Easton文章 http://www.nature.com/ng/journal/vaop/ncurrent/full/ng.3242.html ,研究对象为12万多的欧洲血统女性 。
以前的GWAS研究已经鉴定了79个和乳腺癌相关的位点,这在一定程度上解释了乳腺癌的家族风险。为了鉴定新的易感基因位点,研究人员对11个GWAS研究和41个独立研究进行了Meta分析。这11个GWAS研究共包含了15,748个乳腺癌病例和18,084个对照;而这41个独立的研究包括了46,785个病例和42,892个对照。通过研究其基因结构的微小变化,研究人员确定了15个新的基因位点,这些新的变异与更高的疾病风险有关,研究人员将其被称作单核苷酸多态性(SNPs)。
意味着通过研究已经有总计超过90个与乳腺癌相关的SNPs被发现。该项研究由来自世界各地的数十位乳腺癌协会联合会科学家合作完成,并被发布在2015年的3月9日《自然遗传学》(《Nature Genetics》)上。2017-NG
2017年10月23日,英国《自然》子刊《遗传学》在线发表全球470位科学家的联合研究报告,囊括了剑桥、牛津、哈佛、斯坦福、麻省理工、约翰霍普金斯等331家研究机构,正文共7页,
作者姓名和所在机构名单共5页,研究方法共3页。
在Nature Genetics的文章中,OncoArray联盟分析了全部为欧裔的21,468例 ER阴性患者和100,594例对照以及18,908例 BRCA1突变携带者(含9414例乳腺癌患者)的GWAS数据。结果发现了与ER阴性乳腺癌相关的10个新的变异,同时也验证了另外10个之前报道过的与ER阴性乳腺癌或BRCA1突变携带者相关的变异。这些变异中,多数与ER阴性而不是ER阳性肿瘤有关,显示了这类乳腺癌的特殊遗传特性。
Identification of ten variants associated with risk of estrogen-receptor-negative breast cancer. Nat Genet. 2017 Oct 23. doi: 10.1038/ng.3785.2017-nature
研究人员首先对欧洲和亚洲样本分别进行GWAS研究,采用高密度基因芯片加imputation的方法,一共纳入1100万SNPs。他们先对已经发表的结果进行验证,发现之前报道的102个显著位点中,94个达到显著水平(P<0.05),其中有49个P值小于5×10-8。作者的新发现同样引人注目,他们在65个区域内找到5969个SNPs与乳腺癌的相关性小于5×10-8, 其中9个在亚洲人中得到验证。
OncoArray联盟包括了全球550名研究人员和300家研究机构,研究团队对欧裔122,977 例患者和 105,974例对照以及亚裔14,068 例患者和 13,104例对照进行了GWAS分析,结果发现了65个新的位点与乳腺癌风险增加有关。绝大多数“危险性单核苷酸多态性(SNPs)”都位于基因的远端调控区,而不是编码区。
参考文献
Dwek, M. and Presneau, Nadège (2017) Association analysis identifies 65 new breast cancer risk loci. Nature. ISSN 0028-08362018-使用Hi-C来检查GWAS结果
研究人员通过一种被称为Capture Hi-C(CHi-C)的高通量遗传分析技术,分析了过往全基因组关联研究(GWAS)中发现的乳腺癌相关的63个基因座,将这些基因组中的110个基因与乳腺癌风险增加联系起来,同时发现其中32个基因与乳腺癌存活相关联。该研究结果以《Capture Hi-C identifies putative target genes at 33 breast cancer risk loci》为题在线发表在3月12日的《Nature Communications》上。
ICR的研究人员开发出了高通量、高分辨率高保真的遗传分析技术—CHi-C,以鉴定调控元件与其靶基因间的物理相互作用,这一技术不受调控元件和靶基因的距离限制。他们利用该技术分析了过往GWAS中发现的乳腺癌相关的63个基因座(其中包含大量“基因沙漠”区域),在其中33个基因座中发现了110个乳腺癌风险相关的靶基因,包括94个蛋白质编码基因和16个非编码RNA,其余30个基因区域中未鉴定出特定的基因。
令人惊喜的是,这些基因的大多数在过往的研究中并未与乳腺癌风险相关,这就有效拓宽了乳腺癌风险的基因谱!
参考资料:
Capture Hi-C identifies putative target genes at 33 breast cancer risk lociOncoArray基因分型平台在癌症GWAS应用
肺癌
发表于2017年6月12日的Nature Genetics杂志,利用OncoArray基因分型平台,完成了最大规模的全基因组肺癌易感性研究,涉及样品包括14,803个肺癌样品和12,262个已有数据的欧洲人对照样品,总共有29266个样品和56,450个对照。确定了18个具有全基因组意义的易感基因位点,其中有10个是之前从未发现过的。这些新的基因位点凸显了肺癌亚型:肺腺癌和肺鳞状细胞癌易感模式的显著变化——4个与整体肺癌相关,6个位点与肺腺癌相关。
卵巢癌
来自OncoArray联盟的研究人员对约25500个例被诊断为上皮性卵巢癌的DNA 进行了检测,并对近 41000 名健康人对照组的基因数据进行检测。研究人员准确定位了与罹患上皮性卵巢癌风险相关的 12 种最新遗传变异,包括6个与浆液性卵巢癌相关的位点、2个与粘液性卵巢癌相关的位点以及1个与卵巢子宫内膜样癌相关的位点。
乳腺癌
对比了来自欧洲人群中122,977例乳腺癌患者与105,974例正常对照人群以及东亚人群中14,068例乳腺癌患者与13,104例正常对照人群的基因组,新鉴定出了65个与乳腺癌风险相关的遗传位点。
- Association analysis identifies 65 new breast cancer risk loci
- Identification of ten variants associated with risk of estrogen-receptor-negative breast cancer
因为隐私,原始数据并不公开,但是可以签订协议去申请下载。但是分析后的结果,是可以下载的。http://bcac.ccge.medschl.cam.ac.uk
研究乳腺癌必须知道的两个国际合作组织: - Breast Cancer Association Consortium (BCAC)
- Consortium of Investigators of Modifiers of BRCA1/2 (CIMBA)
做GWAS研究必须了解的两款芯片: - Illumina iCOGS custom array
- OncoArray