前面我们说到了对3784343个的SNP位点来说,3353921个因为人群频率大于了0.05会被过滤掉,还剩下430304值得我好好研究一下。
那么,现在就开始我的表演吧。
首先,看看过滤后那值得探索的43万位点长啥样,如下:
16 84731054 84731054 G T het 165.77 13 60.00 13.81 2 148321842 148321842 C T hom 500.77 16 60.00 31.30 7 91591448 91591448 A G het 206.77 16 60.00 12.92 1 70361880 70361880 A C het 37.77 9 60.00 4.20 21 11185914 11185914 C T het 715.77 209 59.97 3.49 1 194072273 194072273 C A het 635.77 50 60.00 13.25 1 197838902 197838902 C T het 605.77 55 60.00 11.01 18 64898134 64898134 G A het 107.77 21 60.00 5.39 5 94042767 94042767 A G het 211.77 16 60.00 13.24 GL000232.1 30946 30946 G A het 1919.77 189 55.19 10.27
很明显,这些位点已经没有顺序来 ,而且这个格式,勉强来说可以当做是bed格式,但是也不完全一样,要把我们的vcf文件的变异,来根据这个位点文件进行挑选,现有的工具当然是可以做到,比如snpeff等等,但是我懒得去查看那些软件说明书了,自己动手丰衣足食,不就是写一个脚本的事情嘛。
cat snp_filter.hg19_gnomad_genome_filtered snp.vcf |perl -alne '{print if /^#/;print if exists $h{"$F[0]\t$F[1]"} ;$h{"$F[0]\t$F[1]"}=1 if !/:/;}' > snp.filter_gnomAD.vcf
这就是为什么我很讨厌annovar软件的问题,把大家公认的vcf转为自己的格式,害得我还得写代码转回来。
算了。
现在有了这个 snp.filter_gnomAD.vcf 文件,就可以进行snpeff软件的注释咯 , 运行代码很简单,如下:
java -Djava.io.tmpdir=/home/jmzeng/ -Xmx15g -jar ~/biosoft/SnpEff/snpEff/snpEff.jar \ -i vcf GRCh37.75 snp.filter_gnomAD.vcf > snp.filter_gnomAD.snpeff.vcf
位点不多,所以很快就走完了这个流程,其实这个时候注释我反而无法解读,因为snpeff注释的信息太多了,超过一百多种注释信息。但是它给了一个很不错的html报告,可以很清晰的看到这些突变的性质。
先看对这43万位点的一个总结表格吧:
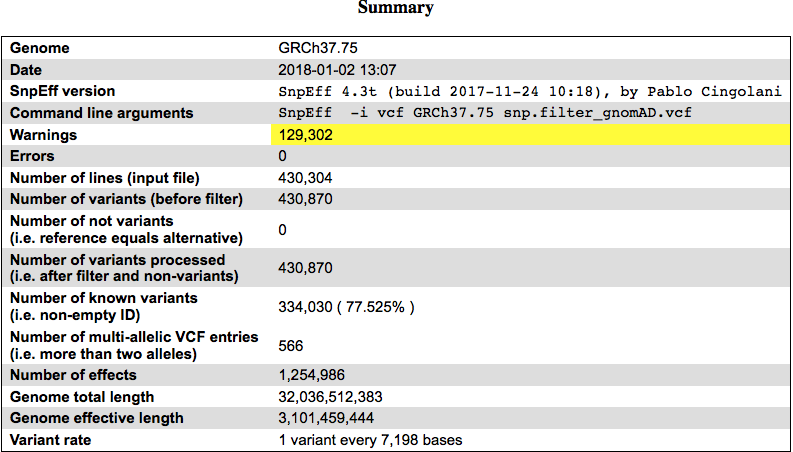
可以看到大部分位点(77.5%)都是在dbSNP数据库里面出现过的,并不是我本人特有的。
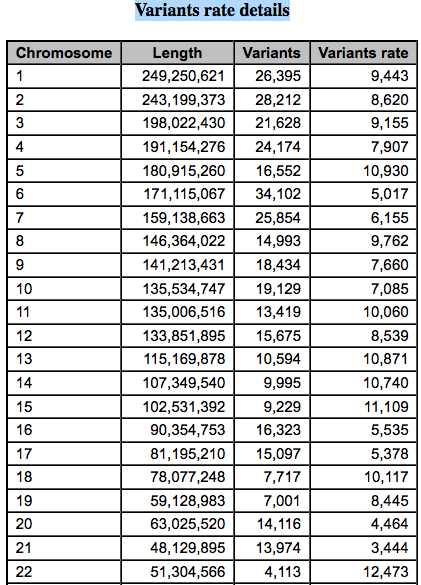
可以看到突变频率还是蛮均一的,至少没有在染色体上面显示出特异性,至于染色体内部嘛,后面的图表再展现即可。
比较奇怪的是chr21和chr22的突变频率相差还是蛮大的,也许值得探究。
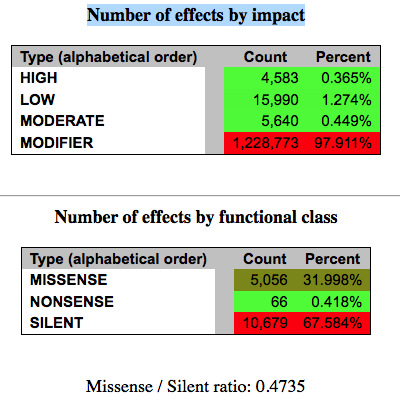
可以看到仍然大部分突变都是silent的,并没有太大的影响,对基因功能产生非常大影响的那些突变才0.365%,当然,就是这么少才值得探究。
但是它们的总数加起来跟43万对不上,应该是还有其它解释。
往期直播目录:
【直播】我的基因组82:如何对maf格式的突变文件统计vaf
从WGS测序得到的VCF文件里面提取位于外显子区域的【直播】我的基因组84
安装snpEFF工具并对VCF文件进行注释【直播】我的基因组85