我是如何学习Gene Fusion分析的
一、Fusion原理
基因融合(Gene fusion)是指将两个或多个基因的编码区首尾相连,置于同一套调控序列(包括启动子、增强子和终止子等)的控制之下,构成嵌合基因。基因融合通常是由于染色体重排所造成的。异常基因融合事件可以引起恶性血液疾病以及肿瘤的发生,所以通过分析基因融合现象将有助于探讨发病机制、biomaker的筛选等,临床意义重大。

二、分析软件列表
Fusion genes/chimeras/translocation finders/structural variations
Genome arrangements result of diseases like cancer can produce aberrant genetic modifications like fusions or translocations. Identification of these modifications play important role in carcinogenesis studies.
- Bellerophontes
- STAR-Fusion
- BreakDancer BreakDancer.
- BreakFusion
- ChimeraScan ChimeraScan.
- DeFuse DeFuse. DeFuse is a software package for gene fusion discovery using RNA-Seq data.
- EBARDenovo EBARDenovo.
- EricScript
- FusionAnalyser FusionAnalyser. FusionAnalyser uses paired reads mapping to different genes (Bridge reads), generated through high-throughput whole transcriptome sequencing, to build a first dataset of candidate fusion events. Subsequently, a second dataset, built upon those reads where only one of the two sequences in a pair is successfully mapped to the reference genome (‘Half-mapped Anchor reads’), is generated. The mapped reads in the latter dataset are used as an anchor to tie each Half-mapped event to the corresponding Bridge region.
- FusionCatcher FusionCatcher. FusionCatcher searches for novel/known somatic fusion genes, translocations, and chimeras in RNA-seq data (stranded/unstranded paired-end reads from Illumina NGS platforms like Solexa/HiSeq/NextSeq/MiSeq) from diseased samples.
- FusionHunter FusionHunter identifies fusion transcripts without depending on already known annotations. It uses Bowtie as a first aligner and paired-end reads.
- FusionMap FusionMap. FusionMap is an efficient fusion aligner which aligns reads spanning fusion junctions directly to the genome without prior knowledge of potential fusion regions. It detects and characterizes fusion junctions at base-pair resolution. FusionMap can be applied to detect fusion junctions in both single- and paired-end dataset from either gDNA-Seq or RNA-Seq studies.
- FusionSeq FusionSeq.
- JAFFA JAFFA is based on the idea of comparing a transcriptome against a reference transcriptome rather than a genome-centric approach like other fusion finders.
- MapSplice
- nFuse
- Oncomine Oncomine - NGS RNA-Seq Gene Expression Browser.
- PRADA prada.
- SOAPFuse SOAPFuse detects fusion transcripts from human paired-end RNA-Seq data. It outperforms other five similar tools in both computation and fusion detection performance using both real and simulated data.
- SOAPfusion Soapfusion.
- TopHat-Fusion TopHat-Fusion is based on TopHat version and was developed to handle reads resulting from fusion genes. It does not require previous data about known genes and uses Bowtie to align continuous reads.
- ViralFusionSeq ViralFusionSeq is high-throughput sequencing (HTS) tool for discovering viral integration events and reconstruct fusion transcripts at single-base resolution. See also hkbic/VFS.
三、软件选择
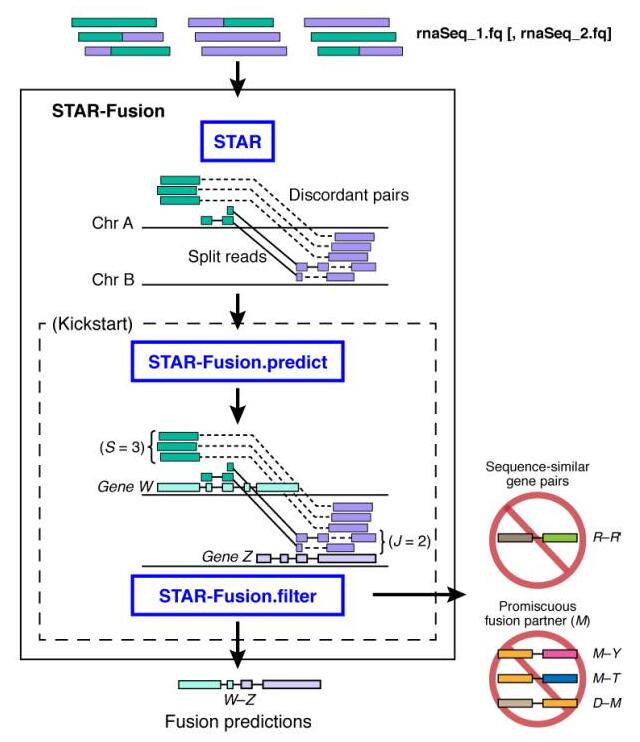
生物信息学鉴定融合转录本的方法一般有两种:①将RNA-seq数据与Reference genome做alignment,鉴别可能发生重排的基因;②先直接将reads装配成更长的转录本序列,再鉴别与重排序列一致的融合转录本。我选择佛-麻省理工学院Broad Institute的 Brian J. Haas 和冷泉港实验室(CSHL)的 Alex Dobin 等人开发的工具STAR-Fusion,其工作原理分为三步:
- ①先将reads通过STAR比对到参考基因组,筛选出split和discordant reads作为候选的融合基因序列;
- ②将候选融合基因序列与参考基因序列进行比对,根据overlaps预测出融合基因;
- ③对预测结果做过滤,去除假阳性结果。
软件下载:
# Get latest STAR source from releases
wget https://github.com/alexdobin/STAR/archive/2.5.3a.tar.gz
tar -xzf 2.5.3a.tar.gz
cd STAR-2.5.3a
# Alternatively, get STAR source using git
git clone https://github.com/alexdobin/STAR.git
cd STAR/source
# Build STAR
make STAR
# To include STAR-Fusion
git submodule update --init --recursive
# If you have a TeX environment, you may like to build the documentation
make manual
----------------------------------------------------------------------------
# Download STAR-Fusion
wget https://github.com/STAR-Fusion/STAR-Fusion/releases/download/v1.1.0/STAR-Fusion_v1.1.0.tar.gz
tar -xzf STAR-Fusion_v1.1.0.tar.gz
新手可以在清华镜像上下载最新版本的Miniconda,命令如下:
wget https://mirrors.tuna.tsinghua.edu.cn/anaconda/miniconda/Miniconda3-4.3.27-Linux-x86_64.sh bash Miniconda3-4.3.27-Linux-x86_64.sh source ~/.bashrc conda config --add channels https://mirrors.tuna.tsinghua.edu.cn/anaconda/cloud/conda-forge/ conda config --add channels https://mirrors.tuna.tsinghua.edu.cn/anaconda/cloud/bioconda conda install STAR conda install STAR-Fusion
Miniconda就像一个手机上的app store,配置好环境之后常用的软件都可以直接简单地下载使用。
四、软件使用
1.STAR的比对分析基本上可以分为两步:一是genomeGenerate(类似于其他比对的软件建立index);二是:序列比对。
①运行genomeGenerate
STAR --runThreadN 12 --runMode genomeGenerate --genomeDir result/ --genomeFastaFiles Homo_sapiens.GRCh38.dna_sm.toplevel.fa --limitGenomeGenerateRAM 146410676608 --sjdbGTFfile Homo_sapiens.GRCh38.90.gtf --sjdbOverhang 89
- --runMode:运行程序模式,默认是比对,所以第一步这个参数设置很关键
- --runThreadN: 运行的线程数
- --genomeDir: 这个参数很重要,是存放你声称index文件路径,需要你事先建立一个有可读写权限的文件夹
- --genomeFastaFiles: 基因组fasta格式文件
- --sjdbGTFfile :GTF注释文件
- --sjdbOverhang: 这个值为你测序read的长度减1,是在注释可变剪切序列的时候使用的最大长度值
详细参数见:https://github.com/alexdobin/STAR/blob/master/doc/STARmanual.pdf
这一步我设置了12个线程还是跑了很久:
等待总是很漫长,尤其是被老板催着要结果的时候。。。
②运行比对
第一步等待的时间很久,等待的过程我也在看比对需要哪些输入,我发现居然可以直接用STAR-Fusion做,直接跳过STAR用原始Fastq数据,简直坑爹,浪费了前面几个小时建立index的时间。。。不过需要提前下载一个genome resource lib,https://data.broadinstitute.org/Trinity/CTAT_RESOURCE_LIB/
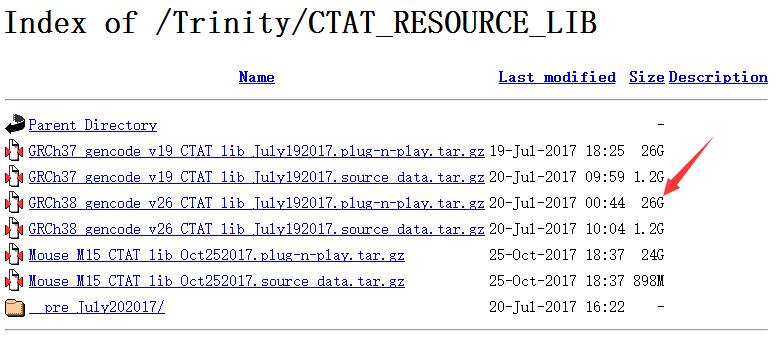
这里的lib有两个版本:GRCh37和GRCh38,每个版本也有两个文件,如果下载1.2G大小的source data,就需要先做一步准备工作:

然后再进行下面直接和下载26个G大小的plug-n-play.tar.gz 一样的,直接用STAR-Fusion运行找融合基因,不需要上面的准备工作。不过26G也够大,让足足我下了一个晚上,中间还断了一次,如果是自己实验室的小型服务器,建议从上面的步骤开始。
STAR-Fusion --genome_lib_dir /path/to/your/CTAT_resource_lib \ --left_fq reads_1.fq \ --right_fq reads_2.fq \ --output_dir star_fusion_outdir
这一步很快,不到15min就跑完了,输出结果:
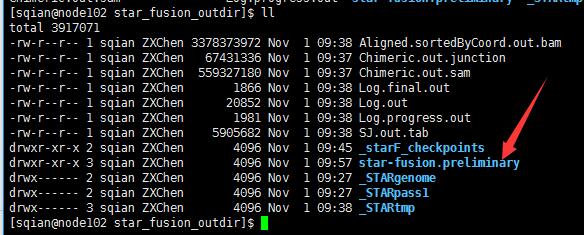
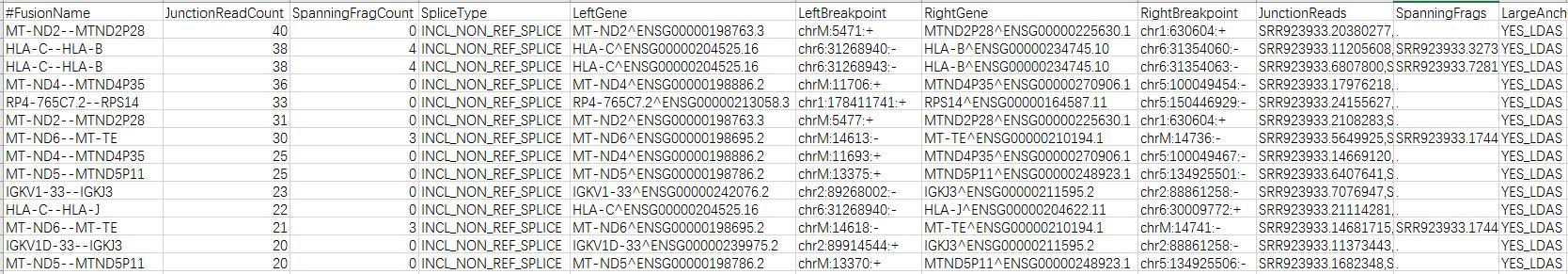
STAR-Fusion详细说明:https://github.com/STAR-Fusion/STAR-Fusion/wiki
大家肯定觉得思路比较混乱,我是按照学习的过程来写的,中间肯定会遇到各种磕磕绊绊,再给大家梳理一下用STAR-Fusion分析融合基因的两种方法:
①先用STAR 建立index,做alignment,再用STAR-Fsion查找融合基因,但要注意在后续下载的genome resource lib和之前alignment的基因组版本对应;
②下载genome resource lib,可以下载1.2G的数据,先做准备工作,再用STAR-Fusion分析,如果网速足够快,可以直接下载26G的数据直接进行分析。
可视化的部分后面再更新。严格来讲,如果我们的数据是RNA-seq产生的,那我们找出来的是融合转录本 ,而不是融合基因 ,如果需要确认是不是融合基因,还需要DNA-Seq的数据。