找变异简单点说,就是把高通量测序得到的成千上万条序列片段比对到合适的参考基因组,找到那些成
功比对的片段与参考基因组的微小差异情况。 那么就涉及到存储测序数据的fastq数据格式,比对的工具,比对后的sam格式,找微小差异的工具,差异结果的vcf文件,每个步骤的软件选择,参数 调整。当然,最重要的是走通整个流程,明白自己在做什么。
一个模拟项目
- 首先下载X,Y染色体的fasta序列,在UCSC上面下载即可。
- 然后把X染色体构建bwa的索引
- 接着模拟一个Y染色体的测序数据,模拟的程序很简单,模拟Y染色体的测序片段(PE100,insert400)
- 然后把模拟测序数据比对到X染色体的参考,统计一下比对结果。
- 最后对比对成功的bam文件进行找变异位点。
代码如下:
## 源代码方式安装 bwa-0.7.15
## conda安装samtools
cd tmp/chrX_Y/hg19/
wget http://hgdownload.cse.ucsc.edu/goldenPath/hg19/chromosomes/chrX.fa.gz
wget http://hgdownload.cse.ucsc.edu/goldenPath/hg19/chromosomes/chrY.fa.gz
gunzip chrX.fa.gz
gunzip chrY.fa.gz
~/biosoft/bwa/bwa-0.7.15/bwa index chrX.fa
perl simulate.pl chrY.fa ## 这个perl脚本在 http://www.bio-info-trainee.com/wp-content/uploads/2015/10/tmp.png
~/biosoft/bwa/bwa-0.7.15/bwa mem -t 5 -M chrX.fa read*.fa >read.sam
samtools view -bS read.sam >read.bam
samtools flagstat read.bam
samtools sort -@ 5 -o read.sorted.bam read.bam
samtools view -h -F4 -q 5 read.sorted.bam |samtools view -bS|samtools rmdup - read.filter.rmdup.bam
samtools index read.filter.rmdup.bam
samtools mpileup -ugf ~/tmp/chrX_Y/hg19/chrX.fa read.filter.rmdup.bam |bcftools call -vmO z -o read.bcftools.vcf.gz
## 把fa/bam/vcf 载入到 IGV 进行可视化,截图其中一个变异位点
## 参考 http://www.biotrainee.com/thread-696-1-1.html
变异寻找的流程
完整的流程可以很复杂:
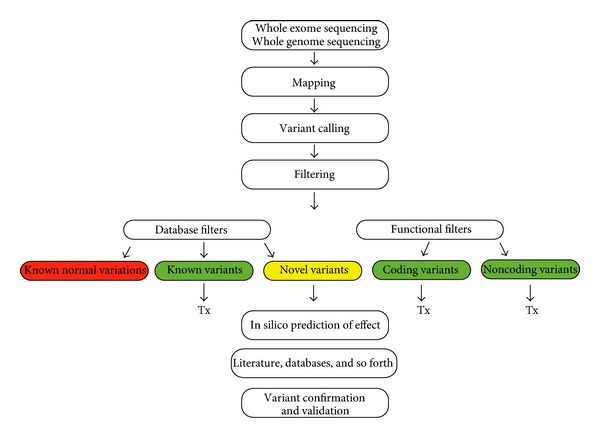
仅是上变异寻找流程就可以很复杂:
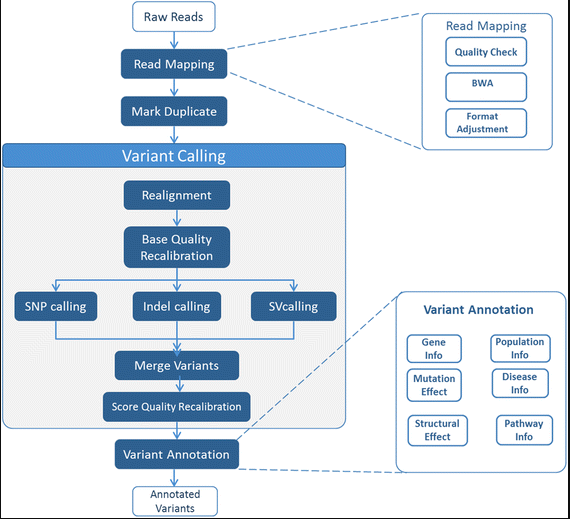
来自于2017年发表于BMC Bioinformatics的文章 MC-GenomeKey: a multicloud system for the detection and annotation of genomic variants