在前面我们提到了用ChIP-seq的分析方法可视化了一下我的WGS数据,结果我们的测序深度分布居然是跟基因组的genomic feature相关的~~~
比如在TSS附近,就很明显看到了一个测序深度峰值,那么前面我们并没有给出直接的解答,而且简单的提了一下这是二代测序的特点,GC含量片段偏好性!
作为一个合格的生物信息学工程师,我当然要把这个理论用自己的代码和数据来亲身实践一遍~
我首先把全基因组的bam文件用mpileup模式输出,根据1000bp的窗口滑动来统计每个窗口的测到的碱基数,GC碱基数,测序总深度!
代码比较复杂,一般人可能理解不来的!
samtools mpileup -f ~/reference/genome/human_g1k_v37/human_g1k_v37.fasta ../P_jmzeng.final.bam|head -1000000 |perl -alne '{$pos=int($F[1]/1000); $key="$F[0]\t$pos";$GC{$key}++ if $F[2]=~/[GC]/;$counts_sum{$key}+=$F[3];$number{$key}++;}END{print "$_\t$number{$_}\t$GC{$_}\t$counts_sum{$_}" foreach sort{$a<=>$b} keys %number}'
而且我代码写的不好,跑10万行需要 4s,跑一百万行需要36s,我估计把这8.9亿行的bam运行完,怎么着也得15个小时。不过不要紧,我们就拿前面的百万行数据做一个测试就好了。
结果如下:
前面两行是窗口的坐标,第几号染色体的第几个窗口,后面3行是数据,分别是每个窗口的测到的碱基数,GC碱基数,测序总深度!
上面的文件就可以导入到R里面进行可视化啦!
PS:这个线性回归图不会看到,自己去搜索哈!(或者去我们生信技能树论坛:http://www.biotrainee.com/thread-695-1-1.html 复制链接到浏览器打开,或者点击阅读原文咯!)
我觉得我这次画的图还不错,很明显能看到这个趋势,GC含量比较高的窗口,有着相应比较高的测序深度!
完美的证明成功啦~
给自己一百个赞,虽然我没有对全基因组数据做验证,但是基因组差异没那么大,我也随机抽样了几次,发现都有这个趋势。
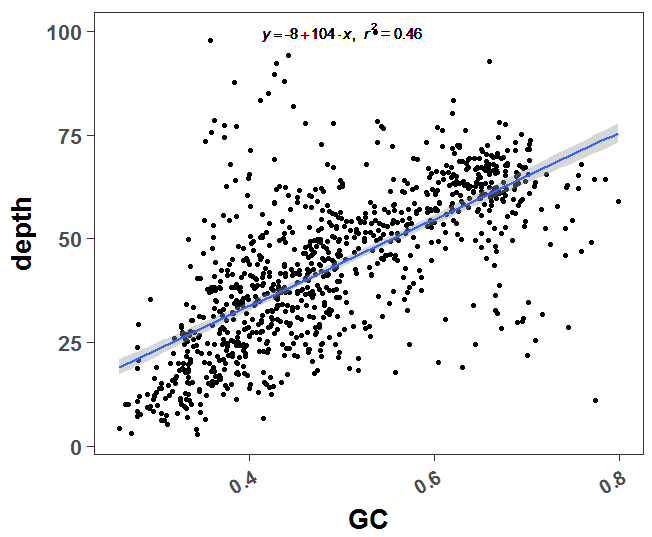
我的R代码如下:
a=read.table('../tmp.txt')
a$GC = a[,4]/a[,3]
a$depth = a[,5]/a[,3]
a = a[a$depth<100,]
plot(a$GC,a$depth)
library(ggplot2)
# GET EQUATION AND R-SQUARED AS STRING
# SOURCE: http://goo.gl/K4yh
lm_eqn <- function(x,y){
m <- lm(y ~ x);
eq <- substitute(italic(y) == a + b %.% italic(x)*","~~italic(r)^2~"="~r2,
list(a = format(coef(m)[1], digits = 2),
b = format(coef(m)[2], digits = 2),
r2 = format(summary(m)$r.squared, digits = 3)))
as.character(as.expression(eq));
}
p=ggplot(a,aes(GC,depth)) + geom_point() +
geom_smooth(method='lm',formula=y~x)+
geom_text(x = 0.5, y = 100, label = lm_eqn(a$GC , a$depth), parse = TRUE)
p=p+theme_set(theme_set(theme_bw(base_size=20)))
p=p+theme(text=element_text(face='bold'),
axis.text.x=element_text(angle=30,hjust=1,size =15),
plot.title = element_text(hjust = 0.5) ,
panel.grid = element_blank(),
#panel.border = element_blank()
)
print(p)