首先上目录:
生信编程很简单↩
人类基因组的外显子区域的长度↩
hg19基因组序列的一些探究↩
hg38每条染色体的基因、转录本分布↩
多个同样行列式文件的合并↩
根据GTF画基因的多个转录本结构↩
下载最新版的KEGG信息,并且解析好↩
写超几何分布检验↩
ID转换↩
根据指定染色体及坐标得到序列↩
根据指定染色体及坐标得到位置信息↩
把文件内容按照染色体分开写出↩
JSON格式数据的格式化↩
多个探针对应一个基因,取平均值、最大值↩
把counts矩阵转换成RPKM矩阵↩
对有临床信息的表达矩阵批量做生存分析↩
对多个差异分析结果直接取交集并集↩
根据GTF格式的基因注释文件得到人所有基因的染色体坐标↩
列出我们板块的人气最旺的20个题目,作为第五章节学习后的复习题目。
生信编程很简单^1
编程语言系统入门
题目
对FASTQ的操作
- 5,3段截掉几个碱基
- 序列长度分布统计
- FASTQ 转换成 FASTA
- 统计碱基个数及GC%
对FASTA的操作
- 取互补序列
- 取反向序列
- DNA to RNA
- 大小写字母形式输出
- 每行指定长度输出序列
- 按照序列长度/名字排序
- 提取指定ID的序列
- 随机抽取序列
高级难度
- 根据坐标取序列
- 多文件合并
- 根据ID列表取序列
- GTF文件探索
- 简并碱基的引物序列还原成多条序列
- snp进行注释并格式化输出
下载安装bowtie2(内含测试数据)
cd ~/biosoft
mkdir bowtie && cd bowtie
wget https://sourceforge.net/projects/bowtie-bio/files/bowtie2/2.2.9/bowtie2-2.2.9-linux-x86_64.zip
unzip bowtie2-2.2.9-linux-x86_64.zip
人类基因组的外显子区域的长度^2
题目
下载人类外显子的坐标文件,编写代码统计外显子区域的长度。
测试数据
- Rbioconductor的TxDb.Hsapiens.UCSC.hg19.knownGene包
- NCBI数据库:ftp://ftp.ncbi.nlm.nih.gov/pub/CCDS/current_human/
R实现代码示例
rm(list=ls())
a=read.table(choose.files(),sep = '\t',stringsAsFactors = F,header = T) # 选择你下的CCDs文件
tmp <- apply(a[1:100,], 1, function(gene){ # 取前100行数据分析调试
# gene=a[3,]
x=gene[10] # Column10 外显子坐标位置列
if(grepl('\\]',x)){ # 判断x中是否存在有]这样的符号,如果有就利用正则替换掉。
x=sub('\\[','',x)
x=sub('\\]','',x)
# 这个时候得到的对象还是像这样的“880073-880179, 880436-880525……”
tmp <- strsplit(as.character(x),',')[[1]]# 我们先从逗号开始分割成小块
start <- as.numeric(unlist(lapply(tmp,function(y){# 取开始位点
strsplit(as.character(y),'-')[[1]][1]
})))
end <- as.numeric(unlist(lapply(tmp,function(y){ # 取结束位点
strsplit(as.character(y),'-')[[1]][2]
})))
gene_d <- data.frame(gene=gene[3], # 将基因名,染色体,开始、结束位点绑定为数据框
chr=gene[1],
start=start,
end=end
)
return (gene_d)#返回数据框
}
})
tmp_pos=c() # 构造一个空的向量
lapply(tmp[1:10], function(x){ # 取前10个list文件计算调试
# print(x)
if ( !is.null(x)){
apply(x, 1,function(y){
#print(y)
for ( i in as.numeric(y[3]):as.numeric(y[4]) ) # y[3]为坐标起点,y[4]为终止坐标,历编
tmp_pos <<- c(tmp_pos,paste0(y[2],"-",i))
})
}
})
length(tmp_pos) # 计算exon的长度
length(unique(tmp_pos)) # 计算去重后的exon的长度
hg19基因组序列的一些探究^3
题目
求:hg19 每条染色体长度,每条染色体N的含量,GC含量。
(高级作业:蛋白编码区域的GC含量会比基因组其它区域的高吗? )
测试数据
- hg19基因组序列下载
wget http://hgdownload.cse.ucsc.edu/goldenPath/hg19/bigZips/chromFa.tar.gz # 也可以在浏览器上下载
tar xvzf chromFa.tar.gz
cat *.fa > hg19.fa
rm chr*.fa # 先把着急删,我待会可能要那他测试运行速度
- 简单测试数据
>chr_1 ATCGTCGaaAATGAANccNNttGTA AGGTCTNAAccAAttGggG >chr_2 ATCGAATGATCGANNNGccTA AGGTCTNAAAAGG >chr_3 ATCGTCGANNNGTAATggGA AGGTCTNAAAAGG >chr_4 ATCGTCaaaGANNAATGANGgggTAPerl代码示例
单行命令 {-}
perl -alne '{if(/^>/){$chr=$_}else{ $A_count{$chr}+=($_=~tr/Aa//); $T_count{$chr}+=($_=~tr/Tt//);$C_count{$chr}+=($_=~tr/Cc//); $G_count{$chr}+=($_=~tr/Gg//); $N_count{$chr}+=($_=~tr/Nn//); }}END{print "$_\t$A_count{$_}\t$T_count{$_}\t$C_count{$_}\t$G_count{$_}\t$N_count{$_}" foreach sort keys %N_count}' test.fa
完整代码 {-}
while (<>){
chomp;
if(/^>/){
$chr=$_
}
else{
$A_count{$chr}+=($_=~tr/Aa//);
$T_count{$chr}+=($_=~tr/Tt//);
$C_count{$chr}+=($_=~tr/Cc//);
$G_count{$chr}+=($_=~tr/Gg//);
$N_count{$chr}+=($_=~tr/Nn//);
}
}
foreach (sort keys %N_count){
$length = $A_count{$_}+$T_count{$_}+$C_count{$_}+$G_count{$_}+$N_count{$_};
$gc = ($G_count{$_}+$C_count{$_})/($A_count{$_}+$T_count{$_}+$C_count{$_}+$G_count{$_});
print "$_\t$A_count{$_}\t$T_count{$_}\t$C_count{$_}\t$G_count{$_}\t$N_count{$_}\t$length\t$gc\n"
}
参考结果{-}
结果如下;
>chr_1 13 10 7 10 4
>chr_2 11 6 5 8 4
>chr_3 10 6 3 10 4
>chr_4 9 4 2 7 3
hg38每条染色体的基因、转录本分布^4
题目
对GTF注释文件进行探究,统计每条染色体基因数、转录本数、内含子数、外显子数。
(高级作业:下载human/rat/mouse/dog/cat/chicken等物种的gtf注释文件(http://asia.ensembl.org/info/data/ftp/index.html),编写函数实现对多个GTF文件进行批量统计染色体基因、转录本的分布及转录本外显子个数;继续探索回答以下问题:所有基因平均有多少个转录本?所有转录本平均有多个exon和intron?如果要比较多个数据库呢(gencode/UCSC/NCBI?)?如果把基因分成多个类型呢?protein coding gene,pseudogene,lncRNA还有miRNA的基因?它们的特征又有哪些变化呢?)
测试数据
wget -c ftp://ftp.ensembl.org/pub/release-87/gtf/homo_sapiens/Homo_sapiens.GRCh38.87.chr.gtf.gz
gzip -d Homo_sapiens.GRCh38.87.chr.gtf.gz
代码示例
# 每条染色体的基因个数
zcat Homo_sapiens.GRCh38.87.chr.gtf.gz |perl -alne '{print if $F[2] eq "gene" }' |cut -f 1 |sort |uniq -c
# 基因分类
zcat Homo_sapiens.GRCh38.87.chr.gtf.gz |perl -alne '{next unless $F[2] eq "gene" ;/gene_biotype "(.*?)";/;print $1}' |sort |uniq -c
多个同样行列式文件的合并^5
题目
将htseq-count生成的所有独立样本文件进行合并(每个样品对应一个文件,包括了所有基因表达量)。
希望通过编程处理每个文件得到输出的表达矩阵(行名是基因名,列名是样品名),如下所示:
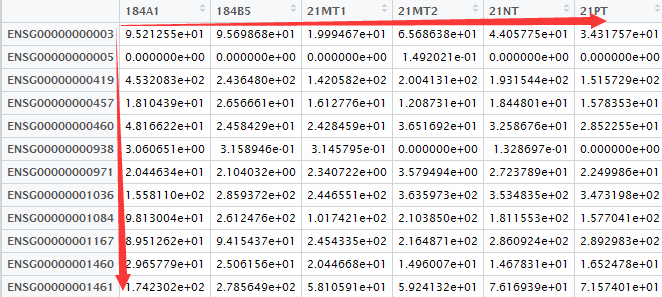
测试数据
模拟数据
用perl脚本模仿htseq-count计算每个样本所有的基因表达量的输出独立样本文件:
## 首先新建文件tmp.sh 输入这个代码:
perl -le '{print "gene_$_\t".int(rand(1000)) foreach 1..99}'
## 然后用perl脚本调用这个tmp.sh文件:
perl -e 'system(" bash tmp.sh >$_.txt") foreach a..z'
## 这样就生成了a~z这26个样本的counts文件
第一列是基因,第二列是该基因的counts值,共有a~z这26个样本的counts文件,需要合并成一个大的行列式,这样才能导入到R里面做差异分析,如果手工用excel表格做,当然是可以的,但是太麻烦,如果有500个样本,正常人都不会去手工做了,需要编程。
每个样本的基因顺序并不一致,这时候你应该怎么做呢?
真实数据
实际需求如下:GSE48213里面有56个文件,需要合并成一个表达矩阵,来根据cell-line的不同,分组做差异分析。可以查看paper
wget -c ftp://ftp.ncbi.nlm.nih.gov/geo/series/GSE48nnn/GSE48213/suppl/GSE48213_RAW.tar
tar -xf GSE48213_RAW.tar
gzip -d *.gz
代码示例
## 首先在GSE48213_RAW目录里面生成tmp.txt文件,使用shell脚本:
awk '{print FILENAME"\t"$0}' * |grep -v EnsEMBL_Gene_ID >tmp.txt
## 然后把tmp.txt导入R语言里面用reshape2处理即可!
setwd('tmp/GSE48213_RAW/')
a=read.table('tmp.txt',sep = '\t',stringsAsFactors = F)
library(reshape2)
fpkm <- dcast(a,formula = V2~V1)
根据GTF画基因的多个转录本结构^6
题目
从NCBI,ENSEMBL,UCSC,GENCODE数据库下载各种GTF注释文件,编写代码得到所有基因的转录本个数,以及每个转录本的外显子的坐标,绘制如下转录本结构图:
![]()
比如对这个ANXA1基因来说,非常多的转录本,但是基因的起始终止坐标,是所有转录本起始终止坐标的极大值和极小值。同时,它是一个闭合基因,因为它存在一个转录本的起始终止坐标等于该基因的起始终止坐标。可以看到它的外显子并不是非常整齐的,虽然多个转录本会共享某些外显子,但是也存在某些外显子比同区域其它外显子长的现象。
测试数据
wget -c http://www.broadinstitute.org/cancer/cga/sites/default/files/data/tools/rnaseqc/gencode.v7.annotation_goodContig.gtf.gz
gzip -d gencode.v7.annotation_goodContig.gtf.gz
R实现代码示例
rm(list=ls())
## http://www.broadinstitute.org/cancer/cga/sites/default/files/data/tools/rnaseqc/gencode.v7.annotation_goodContig.gtf.gz
setwd('tmp')
gtf <- read.table('gencode.v7.annotation_goodContig.gtf.gz',stringsAsFactors = F,
header = F,comment.char = "#",sep = '\t'
)
table(gtf[,2])
gtf <- gtf[gtf[,2] =='HAVANA',]
gtf <- gtf[grepl('protein_coding',gtf[,9]),]
lapply(gtf[1:10,9], function(x){
y=strsplit(x,';')
})
gtf$gene <- lapply(gtf[,9], function(x){
y <- strsplit(x,';')[[1]][5]
strsplit(y,'\\s')[[1]][3]
}
)
draw_gene = 'TP53'
structure = gtf[gtf$gene==draw_gene,]
colnames(structure) =c(
'chr','db','record','start','end','tmp1','tmp2','tmp3','tmp4','gene'
)
gene_start <- min(c(structure$start,structure$end))
gene_end <- max(c(structure$start,structure$end))
tmp_min=min(c(structure$start,structure$end))
structure$new_start=structure$start-tmp_min
structure$new_end=structure$end-tmp_min
tmp_max=max(c(structure$new_start,structure$new_end))
num_transcripts=nrow(structure[structure$record=='transcript',])
tmp_color=rainbow(num_transcripts)
x=1:tmp_max;y=rep(num_transcripts,length(x))
#x=10000:17000;y=rep(num_transcripts,length(x))
plot(x,y,type = 'n',xlab='',ylab = '',ylim = c(0,num_transcripts+1))
title(main = draw_gene,sub = paste("chr",structure$chr,":",gene_start,"-",gene_end,sep=""))
j=0;
tmp_legend=c()
for (i in 1:nrow(structure)){
tmp=structure[i,]
if(tmp$record == 'transcript'){
j=j+1
tmp_legend=c(tmp_legend,paste("chr",tmp$chr,":",tmp$start,"-",tmp$end,sep=""))
}
if(tmp$record == 'exon') lines(c(tmp$new_start,tmp$new_end),c(j,j),col=tmp_color[j],lwd=4)
}
参考结果
下载最新版的KEGG信息,并且解析好^7
题目
下载最新版的KEGG注释文本文件,编写脚本整理成kegg的pathway的ID与基因ID的对应格式。
测试数据
-
1 首先打开KEGG官方网站,网页中展示出了各个物种的分类、拉丁名称、英文名称等信息。
-
2 直接网页中搜索(Ctrl + F)需要下载的物种英文名称或拉丁名。如果不确定物种名称,网站中提供了详细的分类系统,也可根据前面的物种分类信息进行查找。
本文以拟南芥为例,搜索“Arabidopsis thaliana”即可找到。找到后点击物种名称前的3个字母缩写链接(下图红色框中的位置)。

-
3 进入后的网页中包含了物种的一些基因组信息,点击上方的“Brite hierarchy”,进入后再点击“KEGG Orthology (KO)”;
-
4 在跳转出的网页中点击“Download htext”,弹出下载窗口进行下载,国外网站有时会出现无法下载的情况,多试几次即可;

- 5 当然,下载好之后还没有结束。下载得到文本文件,可以看到里面的结构层次非常清楚,C开头的就是kegg的pathway的ID所在行,D开头的就是属于它的kegg的所有的基因。A,B是kegg的分类,总共是6个大类,42个小类。
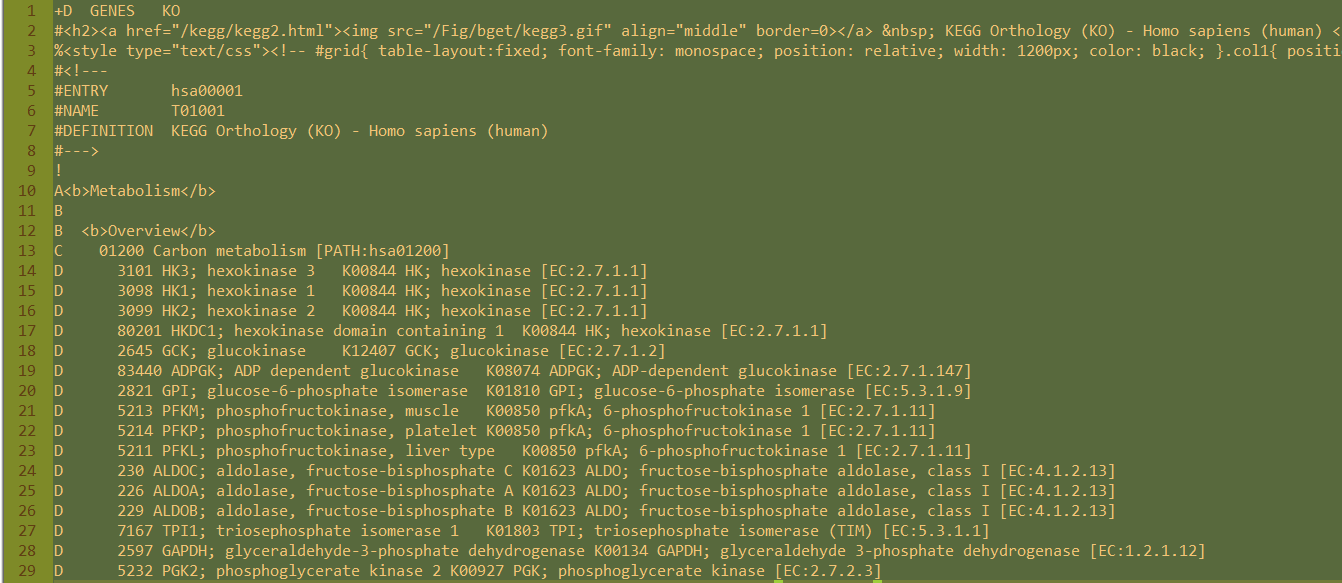
Perl代码示例
perl -alne '{if(/^C/){/PATH:hsa(\d+)/;$kegg=$1}else{print "$kegg\t$F[1]" if /^D/ and $kegg;}}' hsa00001.keg >kegg2gene.txt
参考结果

写超几何分布检验^8
题目
学习GO/KEGG的富集分析的原理,编写代码实现超几何分布检验,将得到的结果与测试数据中的kegg.enrichment.html进行比较。
(P值的计算:C(k,M)*C(n-k,N-M)/C(n,N) )
测试数据
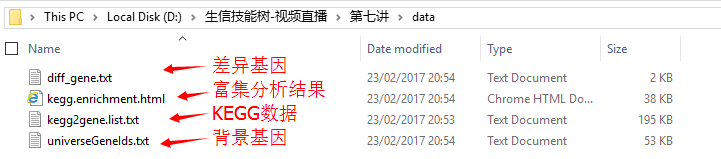
- kegg2gene(第六讲kegg数据解析结果)
暂时不用最新版的kegg注释数据,为了能够统一答案
- 差异基因list和背景基因list(R代码)
source("http://bioconductor.org/biocLite.R")
biocLite("org.Hs.eg.db")
biocLite("KEGG.db")
biocLite("GOstats")
biocLite("hgu95av2.db")
library(org.Hs.eg.db)
library(KEGG.db)
library(GOstats)
library("hgu95av2.db")
##得到kegg2gene.list(KEGG注释信息)
tmp=toTable(org.Hs.egPATH)
write.table(tmp,'kegg2gene.list.txt',quote = F,row.names = F)
## 得到universeGeneIds.txt(背景基因list)
ls('package:hgu95av2.db')
universeGeneIds <- unique(mappedRkeys(hgu95av2ENTREZID))
write.table(universeGeneIds,'universeGeneIds.txt',quote = F,row.names = F)
## 得到diff_gene.txt(差异基因list)
set.seed('123456.789')
diff_gene = sample(universeGeneIds,300)
write.table(diff_gene,'diff_gene.txt',quote = F,row.names = F)
## 得到kegg.enrichment.html(富集分析结果)
annotationPKG='org.Hs.eg.db'
hyperG.params = new("KEGGHyperGParams", geneIds=diff_gene, universeGeneIds=universeGeneIds, annotation=annotationPKG,
categoryName="KEGG", pvalueCutoff=1, testDirection = "over")
KEGG.hyperG.results = hyperGTest(hyperG.params);
htmlReport(KEGG.hyperG.results, file="kegg.enrichment.html", summary.args=list("htmlLinks"=TRUE))
R代码示例
下载链接: https://github.com/jmzeng1314/humanid/blob/master/R/hyperGtest_jimmy.R
library("hgu95av2.db")[/align][align=left]ls('package:hgu95av2.db')
universeGeneIds <- unique(mappedRkeys(hgu95av2ENTREZID))
set.seed('123456.789')
diff_gene = sample(universeGeneIds,300)
library(org.Hs.eg.db)
library(KEGG.db)
tmp=toTable(org.Hs.egPATH)
GeneID2kegg<<- tapply(tmp[,2],as.factor(tmp[,1]),function(x) x)
kegg2GeneID<<- tapply(tmp[,1],as.factor(tmp[,2]),function(x) x)
#results <- hyperGtest_jimmy(GeneID2kegg,kegg2GeneID,diff_gene,universeGeneIds)
GeneID2Path=GeneID2kegg
Path2GeneID=kegg2GeneID
diff_gene_has_path=intersect(diff_gene,names(GeneID2Path))
n=length(diff_gene) #306
N=length(universeGeneIds) #5870
results=c()
for (i in names(Path2GeneID)){
#i="04672"
M=length(intersect(Path2GeneID[[i]],universeGeneIds))
#print(M)
if(M<5)
next
exp_count=n*M/N
#print(paste(n,N,M,sep="\t"))
k=0
for (j in diff_gene_has_path){
if (i %in% GeneID2Path[[j]]) k=k+1
}
OddsRatio=k/exp_count
if (k==0) next
p=phyper(k-1,M, N-M, n, lower.tail=F)
#print(paste(i,p,OddsRatio,exp_count,k,M,sep=" "))
results=rbind(results,c(i,p,OddsRatio,exp_count,k,M))
}
colnames(results)=c("PathwayID","Pvalue","OddsRatio","ExpCount","Count","Size")
results=as.data.frame(results,stringsAsFactors = F)
results$p.adjust = p.adjust(results$Pvalue,method = 'BH')
results=results[order(results$Pvalue),]
rownames(results)=1:nrow(results)
ID转换^9
题目
probe_id,gene_id,gene_name, symbol之间的转换。
测试数据
- 需要转换的探针ID(变量my_probe)
rm(list=ls())
library("hgu95av2.db")
ls('package:hgu95av2.db')
probe2entrezID=toTable(hgu95av2ENTREZID)
probe2symbol=toTable(hgu95av2SYMBOL)
probe2genename=toTable(hgu95av2GENENAME)
my_probe = sample(unique(mappedLkeys(hgu95av2ENTREZID)),30)
tmp1 = probe2symbol[match(my_probe,probe2symbol$probe_id),]
tmp2 = probe2entrezID[match(my_probe,probe2entrezID$probe_id),]
tmp3 = probe2genename[match(my_probe,probe2genename$probe_id),]
write.table(my_probe,'my_probe.txt',quote = F,col.names = F,row.names =F)
write.table(tmp1$symbol,'my_symbol.txt',quote = F,col.names = F,row.names =F)
write.table(tmp2$gene_id,'my_geneID.txt',quote = F,col.names = F,row.names =F)
- 下载对应关系
ftp://ftp.ncbi.nlm.nih.gov/gene/DATA/ - illumina芯片的探针
所有bioconductor支持的芯片平台对应关系:通过bioconductor包来获取所有的芯片探针与gene的对应关系;可以从NCBI的GPL平台下载:http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GPL6947;也可以直接加载对应的包;或者直接去公司的主页下载manifest文件。
library("illuminaHumanv4.db")
ls('package:illuminaHumanv4.db')
probe2entrezID=toTable(illuminaHumanv4ENTREZID)
probe2symbol=toTable(illuminaHumanv4SYMBOL)
probe2genename=toTable(illuminaHumanv4GENENAME)
my_probe = sample(unique(mappedLkeys(illuminaHumanv4ENTREZID)),30)
probe2symbol[match(my_probe,probe2symbol$probe_id),]
probe2entrezID[match(my_probe,probe2entrezID$probe_id),]
probe2genename[match(my_probe,probe2genename$probe_id),]
R代码示例
基因的转换:运行下面的R代码,得到的my_symbol_gene和my_entrez_gene就是需要转换的ID。
library("illuminaHumanv4.db")
ls('package:illuminaHumanv4.db')
my_entrez_gene = sample(unique(mappedRkeys(illuminaHumanv4ENTREZID)),30)
my_symbol_gene = sample(unique(mappedRkeys(illuminaHumanv4SYMBOL)),30)
library("org.Hs.eg.db")
ls('package:org.Hs.eg.db')
entrezID2symbol <- toTable(org.Hs.egSYMBOL)
entrezID2symbol[match(my_entrez_gene,entrezID2symbol$gene_id),]
entrezID2symbol[match(my_symbol_gene,entrezID2symbol$symbol),]
根据指定染色体及坐标得到序列^10
题目
参考基因组hg19,指定染色体及坐标”chr5”,”8397384”,编写程序得到这个坐标以及它上下一个碱基。(机器无法计算hg19,则使用测试数据,指定坐标是 3号染色体的第6个碱基。)
测试数据
>chr_1
ATCGTCGaaAATGAANccNNttGTA
AGGTCTNAAccAAttGggG
>chr_2
ATCGAATGATCGANNNGccTA
AGGTCTNAAAAGG
>chr_3
ATCGTCGANNNGTAATggGA
AGGTCTNAAAAGG
>chr_4
ATCGTCaaaGANNAATGANGgggTA
根据指定染色体及坐标得到位置信息^11
题目
基因的chr,start,end都是已知的(坐标是hg38系统),任意给定基因组的chr:pos(chr1:2075000-2930999), 判断该区间在哪个基因上面?(可用现成软件bedtools)
测试数据
chr7 148697841 148698941
chr7 148698942 148699029
chr7 148699911 148701053
chr7 148701109 148701307
chr7 148701354 148702694
chr7 148703100 148703520
chr7 148703831 148704175
chr7 148704484 148704734
chr7 148704857 148705937
chr7 148706271 148706671
把文件内容按照染色体分开写出^12
题目
根据染色体把文件拆分成1~22和其它染色体的两个文件呢?
测试数据
wget ftp://ftp.ncbi.nlm.nih.gov/pub/CCDS/current_human/CCDS.current.txt
把文件改成bed格式,如下:
chr2 43995310 43995986
chr17 49788603 49789067
chr17 59565573 59566163
chr19 8390308 8390745
chr12 49188033 49189033
chr7 974903 975570
chr7 98878532 98879500
chr7 44044672 44045322
chr1 153634052 153634772
chr11 60905850 60906575
Perl代码示例
use FileHandle;
foreach( 1..22 ){
$normal_chr{"chr".$_}=1 ;
open $fh{"chr".$_},">chr$_.txt" or die;
}
open other,">chr_other.txt" or die;
open FH,'a.bed';
while(<FH>){
chomp;
@F=split;
if(exists $normal_chr{$F[0]}){
$fh{$F[0]}->print( "$_\n" );
}else{
print other $_;
}
}
foreach( 1..22 ){
close $fh{$_};
}
close other;
JSON格式数据的格式化^13
题目
学习json格式,下载测试数据,从该json文件里面提取:technique factor target principal_investigator submission label category type Developmental-Stage organism key这几列信息。
测试数据
http://biotrainee.com/jbrowse/JBrowse-1.12.1/sample_data/json/modencode/modencodeMetaData.json
Perl代码示例
#!/usr/bin/env perl
use strict;
use warnings;
use autodie ':all';
use 5.10.0;
use JSON 2;
my $data = from_json( do { local $/; open my $f, '<', $ARGV[0]; scalar <$f> } );
my @fields = qw( technique factor target principal_investigator submission label category type Developmental-Stage organism key );
say join ',', map "\"$_\"", @fields;
for my $item ( @{$data->{items}} ) {
$item->{key} = $item->{label};
no warnings 'uninitialized';
for my $track ( @{$item->{Tracks}} ) {
$item->{label} = $track;
say join ',', map "\"$_\"", @{$item}{@fields};
}
}
参考结果
完成之后的结果应该是:http://biotrainee.com/jbrowse/JBrowse-1.12.1/sample_data/json/modencode/modencodeMetaData.csv

多个探针对应一个基因,取平均值、最大值 [^14]
[^14]: 多个探针对应一个基因,取平均值、最大值
题目
编写脚本对多个探针对应一个基因,取平均值、最大值。
测试数据
R代码示例
# 平均值
BiocInstaller::biocLite('CLL')
BiocInstaller::biocLite('hgu95av2.db')
library('hgu95av2.db')
library(CLL)
data(sCLLex)
sCLLex=sCLLex[,1:8] ## 样本太多,我就取前面8个
group_list=sCLLex$Disease
exprSet=exprs(sCLLex)
exprSet=as.data.frame(exprSet)
exprSet$probe_id=rownames(exprSet)
head(exprSet)
probe2symbol=toTable(hgu95av2SYMBOL)
dat=merge(exprSet,probe2symbol,by='probe_id')
results=t(sapply(split(dat,dat$symbol),function(x) colMeans(x[,1:(ncol(x)-1)])))
把counts矩阵转换成RPKM矩阵[^15]
[^15]: 把counts矩阵转换成RPKM矩阵
题目
编写脚本将hisat2+htseq-counts之后得到reads的counts矩阵转换成RPKM矩阵。
测试数据
wget ftp://ftp.ncbi.nlm.nih.gov/pub/CCDS/current_mouse/CCDS.current.txt
grep -v '^#' CCDS.current.txt | perl -alne '{/\[(.*?)\]/;$len=0;foreach(split/,/,$1){@tmp=split/-/;$len+=($tmp[1]-$tmp[0])};$h{$F[2]}=$len if $len >$h{$F[2]}} END{print "$_\t$h{$_}" foreach sort keys %h}' >mm10_ccds_length.txt
R代码示例
genes_len=read.table("mm10_ccds_length.txt",stringsAsFactors=F)
head(genes_len)
V1 V2
1 -343C11.2 1139
2 00R_AC107638.2 1060
3 00R_Pgap2 1676
4 0610005C13Rik 7363
5 0610006L08Rik 34995
6 0610007P14Rik 9074
colnames(genes_len)<- c("GeneName","Len")
head(exprSet)
GSM860181_priSG-A GSM860182_SG-A GSM860183_SG-B GSM860184_lepSC
00R_AC107638.2 0 1 0 1
0610005C13Rik 20 22 11 27
0610006L08Rik 0 0 0 2
0610007P14Rik 2075 1785 1269 1430
0610009B22Rik 256 376 300 386
0610009E02Rik 22 22 16 28
exprSet<-exprSet[ rownames(exprSet) %in% genes_len$GeneName ,]
total_count<- colSums(exprSet)
neededGeneLength=genes_len[ match(rownames(exprSet), genes_len$GeneName) ,2]
rpkm <- t(do.call( rbind,lapply(1:length(total_count),function(i){
10^9*exprSet[,i]/neededGeneLength/total_count[i]
}) ))
head(rpkm)
rownames(rpkm)=rownames(exprSet)
colnames(rpkm)=colnames(exprSet)
write.table(rpkm,file="rpkm.txt",sep="\t",quote=F)
library(TxDb.Mmusculus.UCSC.mm10.knownGene)
txdb=TxDb.Mmusculus.UCSC.mm10.knownGene
gt=transcriptsBy(txdb,by="gene")
lapply(gt[1:40],function(x) max(width(x)))
library(org.Mm.eg.db)
mykeys=
columns(txdb);keytypes(txdb)
neededcols <- c("GENEID", "TXSTRAND", "TXCHROM")
select(txdb, keys=mykeys, columns=neededcols, keytype="TXNAME")
参考结果
对有临床信息的表达矩阵批量做生存分析^16
题目
使用R实现生存分析:
用my.surv <- surv(OS_MONTHS,OS_STATUS=='DECEASED')构建生存曲线;
用kmfit2 <- survfit(my.surv~TUMOR_STAGE_2009)来做某一个因子的KM生存曲线;
用survdiff(my.surv~type, data=dat)来看看这个因子的不同水平是否有显著差异,其中默认用是的logrank test 方法;
用coxph(Surv(time, status) ~ ph.ecog + tt(age), data=lung)来检测自己感兴趣的因子是否受其它因子(age,gender等等)的影响。
代码示例
rm(list=ls())
## 50 patients and 200 genes
dat=matrix(rnorm(10000,mean=8,sd=4),nrow = 200)
rownames(dat)=paste0('gene_',1:nrow(dat))
colnames(dat)=paste0('sample_',1:ncol(dat))
os_years=abs(floor(rnorm(ncol(dat),mean = 50,sd=20)))
os_status=sample(rep(c('LIVING','DECEASED'),25))
library(survival)
my.surv <- Surv( os_years,os_status=='DECEASED')
## The status indicator, normally 0=alive, 1=dead. Other choices are TRUE/FALSE (TRUE = death) or 1/2 (2=death).
## And most of the time we just care about the time od DECEASED .
fit.KM=survfit(my.surv~1)
fit.KM
plot(fit.KM)
log_rank_p <- apply(dat, 1, function(values1){
group=ifelse(values1>median(values1),'high','low')
kmfit2 <- survfit(my.surv~group)
#plot(kmfit2)
data.survdiff=survdiff(my.surv~group)
p.val = 1 - pchisq(data.survdiff$chisq, length(data.survdiff$n) - 1)
})
names(log_rank_p[log_rank_p<0.05])
gender= ifelse(rnorm(ncol(dat))>1,'male','female')
age=abs(floor(rnorm(ncol(dat),mean = 50,sd=20)))
## gender and age are similar with group(by gene expression)
cox_results <- apply(dat , 1, function(values1){
group=ifelse(values1>median(values1),'high','low')
survival_dat <- data.frame(group=group,gender=gender,age=age,stringsAsFactors = F)
m=coxph(my.surv ~ age + gender + group, data = survival_dat)
beta <- coef(m)
se <- sqrt(diag(vcov(m)))
HR <- exp(beta)
HRse <- HR * se
#summary(m)
tmp <- round(cbind(coef = beta, se = se, z = beta/se, p = 1 - pchisq((beta/se)^2, 1),
HR = HR, HRse = HRse,
HRz = (HR - 1) / HRse, HRp = 1 - pchisq(((HR - 1)/HRse)^2, 1),
HRCILL = exp(beta - qnorm(.975, 0, 1) * se),
HRCIUL = exp(beta + qnorm(.975, 0, 1) * se)), 3)
return(tmp['grouplow',])
})
cox_results[,cox_results[4,]<0.05]
PS: 里面的os_years应该修改为os_month,因为总的生存期不应该长达几十年,而且因为ag和os_years都是随机生成的,可能会出现很不符合自然科学的现象。但是这个是模拟数据,请大家不用较真。
对多个差异分析结果直接取交集并集[^17]
[^17]: 对多个差异分析结果直接取交集并集
题目
编写脚本对每两个差异分析结果计算基因交集个数与基因并集个数的比值,得到一个比值矩阵。
测试数据
用perl单行命令模拟数据:
perl -e 'BEGIN{use List::Util qw/shuffle/;@gene=A..Z}{foreach(1..10){@this_genes=@gene[(shuffle(0..25))[0..int(rand(20))+4]];print "comparison_$_ <- ";print join(";",@this_genes);print "\n"}}'
总共10次差异分析,每次差异分析得到的差异基因在同一行的后面用大小字母表示。
comparison_1 -> I;G;E;V;C;K;B;W
comparison_2 -> G;E;N;H;Y;M;L;S;K;A;J;O;D;P;R;U;Q;F;Z;C
comparison_3 -> Y;V;U;N;H;K;I;P;S;F;D;X;G;C;Z;J;Q;T;W;O;E;M
comparison_4 -> N;T;K;B;H;Z;W;C;Q;I;V;F;D;S;R;Y;J;X;P;O;G;L;A
comparison_5 -> G;J;A;H;W;T;Z;E;Y;S;R
comparison_6 -> Z;M;D;R;P;G;L;W;Y;U;X;E;A;S;T;I;H
comparison_7 -> H;Z;T;O;W;Q;M;X;J;N;U;K;F;P;I;C;S;Y;A;B
comparison_8 -> A;R;L;T;W;Q;S;F;P;X;E;V;Y;G;K;J;Z;C
comparison_9 -> J;X;K;D;O;H;L;F;C;P;R;N
comparison_10 -> G;S;K;H;C;O;W;F;Q;X
R代码示例
a=readLines('test.txt')
n=unlist(lapply(a , function(x){
trimws(strsplit(x,'<-')[[1]][1])
}))
v=lapply(a , function(x){
trimws(strsplit(trimws(strsplit(x,'<-')[[1]][2]),';')[[1]])
})
names(v)=n
tmp=unlist(lapply(v, function(x){
lapply(v, function(y){
p1=length(intersect(x,y))
p2=length(union(x,y))
return(p1/p2)
})
}))
out=matrix(tmp,nrow = length(n))
rownames(out)=n
colnames(out)=n
out[1:5,1:5]
参考结果
结果的前五行如下:
comparison_1 comparison_2 comparison_3 comparison_4 comparison_5
comparison_1 1.0000000 0.1666667 0.3043478 0.2916667 0.1875000
comparison_2 0.1666667 1.0000000 0.6800000 0.6538462 0.4090909
comparison_3 0.3043478 0.6800000 1.0000000 0.7307692 0.3750000
comparison_4 0.2916667 0.6538462 0.7307692 1.0000000 0.4166667
comparison_5 0.1875000 0.4090909 0.3750000 0.4166667 1.0000000
根据GTF格式的基因注释文件得到人所有基因的染色体坐标[^18]
[^18]: 根据GTF格式的基因注释文件得到人所有基因的染色体坐标
题目
从gencode数据库里面可以下载所有的gtf文件,编写脚本得到基因的染色体、起始终止坐标如下:
[jianmingzeng@gencode]$ head protein_coding.hg19.position
chr1 69091 70008 OR4F5
chr1 367640 368634 OR4F29
chr1 621096 622034 OR4F16
chr1 859308 879961 SAMD11
chr1 879584 894689 NOC2L
chr1 895967 901095 KLHL17
chr1 901877 911245 PLEKHN1
chr1 910584 917473 PERM1
chr1 934342 935552 HES4
chr1 936518 949921 ISG15
[jianmingzeng@gencode]$ head protein_coding.hg38.position
chr1 69091 70008 OR4F5
chr1 182393 184158 FO538757.2
chr1 184923 200322 FO538757.1
chr1 450740 451678 OR4F29
chr1 685716 686654 OR4F16
chr1 923928 944581 SAMD11
chr1 944204 959309 NOC2L
chr1 960587 965715 KLHL17
chr1 966497 975865 PLEKHN1
chr1 975204 982093 PERM1
名词解释 {#glossary}
包括测序相关,组学相关的,给第1,6章节作补充。