首先在UCSC的table browser 里面下载下面这个文件:
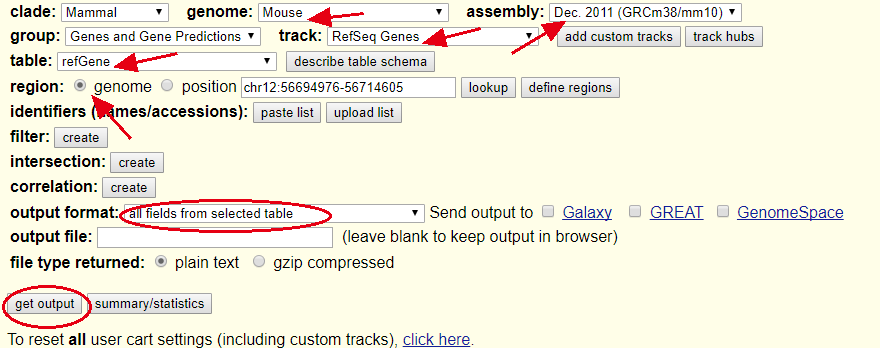
可以看到我这里选择的mm10的refseq系统的所有基因,共有29037个不同的tss,36872个转录本,只有24540个基因,说明有部分基因有多个tss,这个其实挺麻烦的。
#bin name chrom strand txStart txEnd cdsStart cdsEnd exonCount exonStarts exonEnds score name2 cdsStartStat cdsEndStat exonFrames
0 NM_001282945 chr1 - 134199214 134235457 134202950 134234355 3 134199214,134234014,134235227, 134203590,134234446,134235457, 0 Adora1 cmpl cmpl 2,0,-1,
0 NM_001039510 chr1 - 134199214 134235457 134202950 134234355 3 134199214,134234014,134235227, 134203590,134234412,134235457, 0 Adora1 cmpl cmpl 2,0,-1,
0 NM_001291930 chr1 - 134199214 134235457 134202950 134203505 2 134199214,134235227, 134203590,134235457, 0 Adora1 cmpl cmpl 0,-1,
0 NM_001291928 chr1 - 134199214 134234856 134202950 134234733 2 134199214,134234662, 134203590,134234856, 0 Adora1 cmpl cmpl 2,0,
0 NM_001008533 chr1 - 134199214 134235457 134202950 134234355 2 134199214,134234014, 134203590,134235457, 0 Adora1 cmpl cmpl 2,0,
其实里面可以设置直接下载所有基因的TSS区域的bed文件,可是我不会设置各种参数,也懒得去摸索,直接对上面的文件我可以写脚本处理得到需要的数据形式。
需要输出的是bed格式文件,如下:
chrom / chromStart /chromEnd /name /score /strand
我这里定义的TSS(转录起始位点)区域上下游2.5kb,所以代码如下:
perl -alne '{next if /^#/;if($F[3] eq "+"){$start=$F[4]-2500;$end=$F[4]+2500}else{$start=$F[5]-2500;$end=$F[5]+2500}print join("\t",$F[2],$start,$end,$F[12],0,$F[3])}' ucsc.refseq.txt |sort -u >ucsc.refseq.tss.bed
最后得到的文件如下:
tail ucsc.refseq.tss.bed
chrY 816212 821212 Uba1y 0 +
chrY 81798997 81803997 Gm20747 0 -
chrY 82222714 82227714 Gm20736 0 +
chrY 83925411 83930411 Gm20854 0 -
chrY 85527019 85532019 Gm20854 0 -
chrY 8832669 8837669 Gm20815 0 -
chrY 895287 900287 Kdm5d 0 +
chrY 90752550 90757550 G530011O06Rik 0 -
chrY 90782941 90787941 Erdr1 0 +
chrY 90836906 90841906 G530011O06Rik 0
记住,这个时候,部分基因还有多个tss哦。