最近在写一篇很有趣的文章,一张图说清楚wgs,wes,rna-seq,chip-seq的异同点!
需要用到一些测试数据,我准备拿17号染色体的40437407-40486397这约48Kb碱基区域来举例子,就需要把这个区域的bam提取出来。
我分别找了以前处理的wgs,wes,rna-seq,chip-seq公共数据,原始bam非常大,尤其是WGS的,45G的bam文件,所以只能抽取17号染色体的40437407-40486397这约48Kb碱基区域,以前我做mpileup或者其它都是用的-r 参数,所以我想当然的使用下面的代码:
samtools view -h -r chr17:40437407-40486397 your.sorted.merge.bam |samtools view -bS - >wes.bam
发现始终不对,让我着实郁闷,我就Google了一下,https://www.biostars.org/p/48719/
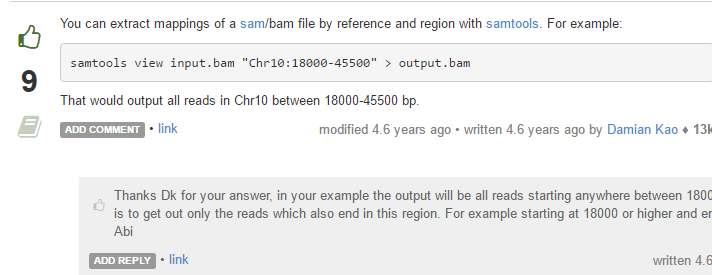
才明白,samtools的view命令的-r参数不再是用来指定坐标了!
samtools view -h control_1.sort.bam "chr17:40437407-40486397" |samtools view -bS - >RNA-seq.bam
所以我修改了命令,完成了提取指定区域比对的reads的bam文件这个需求!
samtools view -h
Usage: samtools view [options] <in.bam>|<in.sam>|<in.cram> [region ...]
Options:
-b output BAM
-C output CRAM (requires -T)
-1 use fast BAM compression (implies -b)
-u uncompressed BAM output (implies -b)
-h include header in SAM output
-H print SAM header only (no alignments)
-c print only the count of matching records
-o FILE output file name [stdout]
-U FILE output reads not selected by filters to FILE [null]
-t FILE FILE listing reference names and lengths (see long help) [null]
-L FILE only include reads overlapping this BED FILE [null]
-r STR only include reads in read group STR [null]
-R FILE only include reads with read group listed in FILE [null]
-q INT only include reads with mapping quality >= INT [0]
-l STR only include reads in library STR [null]
-m INT only include reads with number of CIGAR operations consuming
query sequence >= INT [0]
-f INT only include reads with all bits set in INT set in FLAG [0]
-F INT only include reads with none of the bits set in INT set in FLAG [0]
-x STR read tag to strip (repeatable) [null]
-B collapse the backward CIGAR operation
-s FLOAT integer part sets seed of random number generator [0];
rest sets fraction of templates to subsample [no subsampling]
-@, --threads INT
number of BAM/CRAM compression threads [0]
-? print long help, including note about region specification
-S ignored (input format is auto-detected)
--input-fmt-option OPT[=VAL]
Specify a single input file format option in the form
of OPTION or OPTION=VALUE
-O, --output-fmt FORMAT[,OPT[=VAL]]...
Specify output format (SAM, BAM, CRAM)
--output-fmt-option OPT[=VAL]
Specify a single output file format option in the form
of OPTION or OPTION=VALUE
-T, --reference FILE
Reference sequence FASTA FILE [null]