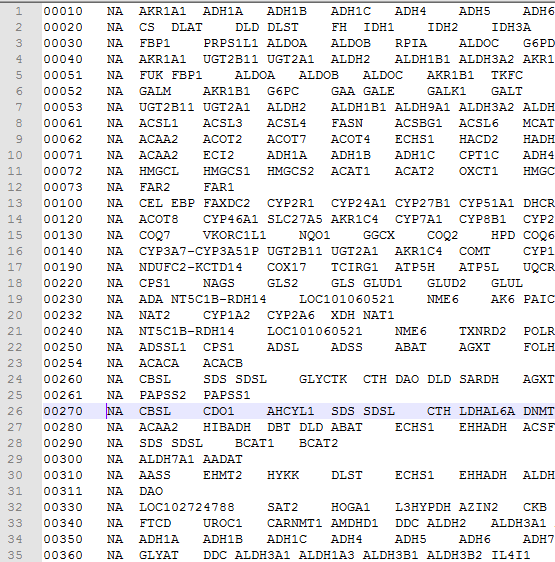
熟悉GSEA软件的都知道,它只需要GCT,CLS和GMT文件,其中GMT文件,GSEA的作者已经给出了一大堆!就是记录broad的Molecular Signatures Database (MSigDB) 已经收到了18026个geneset,但是我奇怪的是里面竟然没有包括cancer testis的gene set,MSigDB的确是多,但未必全,其实里面还有很多重复。而且有不少几乎没有意义的gene set。那我想做自己的gene set来用gsea软件做分析,就需要自己制造gmt格式的数据。因为即使下载了MSigDB的gene set,本质上就是gmt格式的数据而已:http://software.broadinstitute.org/cancer/software/gsea/wiki/index.php/Data_formats#GMT:_Gene_Matrix_Transposed_file_format_.28.2A.gmt.29

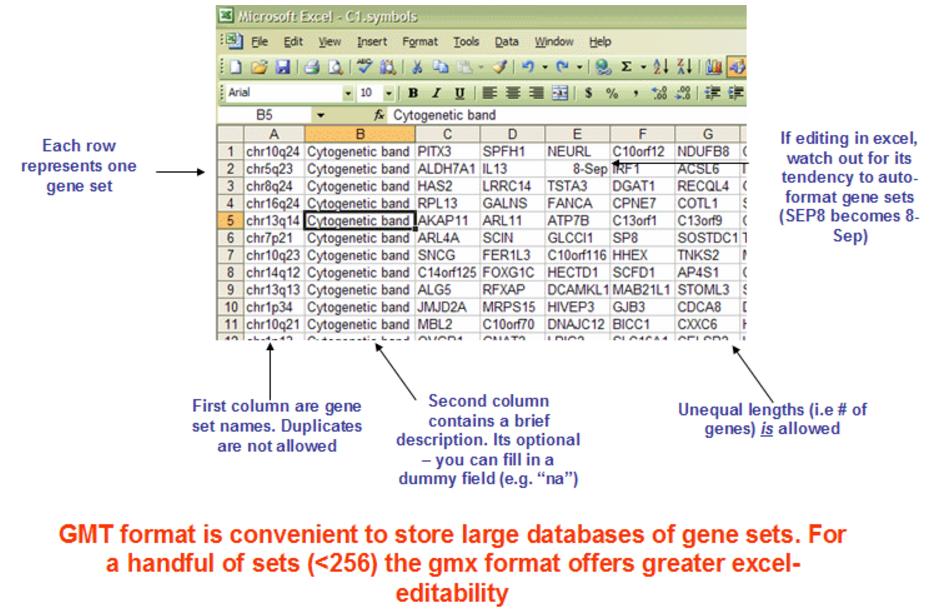
我们首先要拿到自己感兴趣的gene set里面的gene list,最好是以hugo规定的标准symbol。
比如我感兴趣的是 :http://www.cta.lncc.br/modelo.php
我这里提供一个2列的文件,直接转换成gmt的R代码!
文件来自于:下载最新版的KEGG信息,并且解析好,如下:

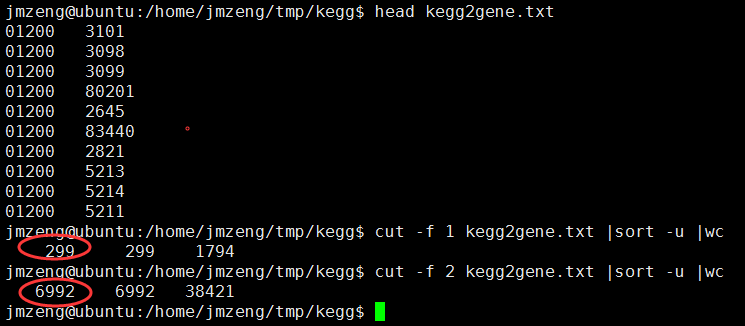
首先在R里面赋值一个变量path2gene_file就是图中的kegg2gene.txt文件,读到R里面去
tmp=read.table(path2gene_file,sep="\t",colClasses=c('character'))
#tmp=toTable(org.Hs.egPATH)
# first column is kegg ID, second column is entrez ID
GeneID2kegg_list<<- tapply(tmp[,1],as.factor(tmp[,2]),function(x) x)
kegg2GeneID_list<<- tapply(tmp[,2],as.factor(tmp[,1]),function(x) x)
这个变量kegg2GeneID_list是一个list,因为是entrez gene ID,需要转换成symbol,我就不多说了,转换后的数据,就是kegg2symbol_list 。

最后对 kegg2symbol_list 输出成gmt文件:
write.gmt <- function(geneSet=kegg2symbol_list,gmt_file='kegg2symbol.gmt'){sink( gmt_file )for (i in 1:length(geneSet)){cat(names(geneSet)[i])cat('\tNA\t')cat(paste(geneSet[[i]],collapse = '\t'))cat('\n')}sink()}