看了illumina的测序仪市场份额的确很夸张,像我这样在生信数据分析领域身经百战的老鸟,都是直到今天才碰到color space的测序数据。测序平台是AB 5500xl Genetic Analyzer,就是传说中的solid格式。主要是我在学习一篇关于tp53转录因子结合能力的文章的时候碰到的 ,我查看了下载的数据虽然还是fastq格式,但很诡异,我完全不认识里面的序列。这里总结一下,下面是我的学习过程及思路,有点乱,大家随便看看!
首先:测序仪给的数据应该是 (.csfasta & .qual) 这两个后缀名的文件
然后,可以用脚本把数据转为csfastq格式, 与普通fastq数据格式是没有区别,但是里面包含的不是序列,是color的编码。
其次,color space不允许转为base space数据!!!
最后,之所以转为csfastq格式,是为了适应很多软件,fastqc,cutadap,SHRiMP,sequel和BFAST ,bowtie等等
csfastq数据如下,还是四行代表一条read:
@SRR2967009.1 100_1000_1168_F3T10011023211201220121202030102221012302121010131001+@SRR2967009.2 100_1000_1211_F3T20132312201120021312220200023110220113100012321011+@@@@@@@@@<@@@@@@@@@@@@@@@@@@@@@@?@@@@/?@@@@@@@@<?@@SRR2967009.3 100_1000_1272_F3T33222002231020000110132110001032232200332111022002
起初,我完全蒙圈了,查了资料才勉强了解。
Generally, in a classic fastq format file, first line is begin with "@", 2nd line is the sequence of reads, 3rd line is a "+" and 4th line is the quality.
However in these fastq files, the sequence of reads are some numbers ("0,1,2,3").
However in these fastq files, the sequence of reads are some numbers ("0,1,2,3").
其实这个fastq并不是测序仪的下机数据,测序仪给的数据应该是 (.csfasta & .qual) 这两个后缀名的文件,一般情况下我们需要把SOLid output files (.csfasta & .qual) into an integrated .csfastq file,转为的csfastq就是fastq格式了,但是跟通常的fastq有略微区别
所以我们的fastq里面的不是序列,而是color的编码,Colors may be encoded either as numbers (
0=blue, 1=green, 2=orange, 3=red) or as characters A/C/G/T (A=blue, C=green, G=orange, T=red).>1_53_33_F3
T2213120002010301233221223311331
>1_53_70_F3
T2302111203131231130300111123220
...Here, T is the primer base. bowtie detects and handles primer bases properly (i.e., the primer base and the adjacent color are both trimmed away prior to alignment) as long as the rest of the read is encoded as numbers.
如果从sra数据库里面下载数据的时候知道是solid的数据,就应该用abi-dump而不是fastq-dump
首先下载数据并且解压:
for ((i=7009;i<7014;i++)) ;do wget ftp://ftp-trace.ncbi.nlm.nih.gov/sra/sra-instant/reads/ByStudy/sra/SRP/SRP066/SRP066824/SRR296$i/SRR296$i.sra;done
因为测序平台是AB 5500xl Genetic Analyzer,就是传说中的solid格式,所以不应该用fastq-dump啦,应该用abi-dump才对!
ls *sra |while read id; do ~/biosoft/sratoolkit/sratoolkit.2.6.3-centos_linux64/bin/abi-dump $id;done
解压之后是下面这样:

这样只能转为csfasta格式文件和qual文件,需要下载大名鼎鼎的lh写的一个脚本:wget http://www.bbmriwiki.nl/svn/bwa_45_patched/solid2fastq.pl 来转为fastq格式
程序非常好用:perl solid2fastq.pl SRR2967009_ SRR2967009 即可
也可以用Python程序来做这个转换,http://edison.cremag.org/resources/seq-analysis/tools/solid2fastq/
最后就是输出了fastq格式的 color space的数据 ,但是我测试了,直接用fastq-dump也可以把数据解压成fastq格式的color space的数据,并不需要那么麻烦的, 因为我们不是从测序仪拿数据,而是从SRA数据库里面直接下载。(补充一下,直接用fastq-dump也可以把数据解压成fastq格式跟用abi-dump解压后再转换成csfastq有区别,但是我现在说不清楚区别是什么,建议用abi-dump)
SOLiD native (CSFASTA/QUAL)
All SRA data can be output into color space data. The utility ‘abi-dump’ can be used to output CSFASTA and QUAL data files (with appropriate options, fastq-dump can be used to output “CSFASTQ” format).
SHRiMP,sequel和BFAST 都可以来比对fastq格式的color space的数据,或者直接从 (.csfasta & .qual) 这两个文件开始处理,其实bowtie也可以的。
比对后的bam文件,就可以走正常的illumina数据分析流程啦!
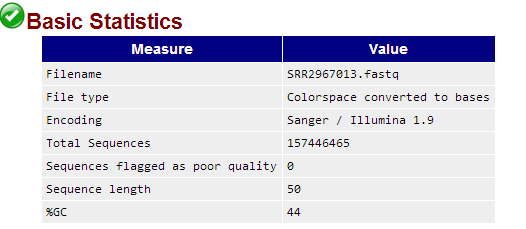
转为了fastq格式的color space的数据,就可以直接进行fastqc看看质量控制图片,如果质量很差,可以直接用处理cutadapt等各种软件进行处理,in a
.csfasta and a .qual file (this is the native SOLiD format).fastqc软件直接处理csfastq格式数据结果如下:

Sequencer reads have a chance of read error (e.g. spot misidentification), combined with a chance of sequence error (e.g. polymerase misread in the PCR step).
For sequencers that output in base space, both these errors have a similar effect on the base-space mapping.
For sequencers that output in color-space, the read errors result in a somewhat unexpected base-space translation even if the underlying sequence has a perfect match to the reference.
The issues relating to color-space to base-space translation were discussed in the thread you linked to, but here's my take on it (dumped from an email I recently sent to someone else):A color-space sequence is an encoding of adjacent dimers such that unchanging bases are encoded with '0', complementary changes are encoded with '3', the colour '1' is used for a non-complementary base change on the same side of the alphabet (AC, CA, GT, or TG), and the colour '2' is used for a non-complementary base change on a different side of the alphabet (AG, GA, CT, or TC). A table of these changes can be found here:
http://www.ploscompbiol.org/article/...i.1000386.g002
This has a few nice properties (e.g. the reverse-complement of a color-space sequence is the same as the reverse of the color-space sequence, a SNP will have two transitions), but many annoying and nasty properties.
http://www.ploscompbiol.org/article/...i.1000386.g002
This has a few nice properties (e.g. the reverse-complement of a color-space sequence is the same as the reverse of the color-space sequence, a SNP will have two transitions), but many annoying and nasty properties.
The first is that a color-space sequence in itself is meaningless without a base reference (usually the starting base).