VEP是国际三大数据库之一的ENSEMBL提供的,也是非常主流和方便,但它是基于perl语言的,所以在模块方面可能会有点烦人。跟snpEFF一样,也是对遗传变异信息提供更具体的注释,而不仅仅是基于位点区域和基因。如果你熟悉外显子联盟这个数据库EXAC(ExAC.r0.3.sites.vep.vcf.gz),你可以下载它所有的突变记录数据,看看它对每个变异位点到底注释了些什么,它就是典型的用VEP来注释的。 Continue reading
五
05
VEP是国际三大数据库之一的ENSEMBL提供的,也是非常主流和方便,但它是基于perl语言的,所以在模块方面可能会有点烦人。跟snpEFF一样,也是对遗传变异信息提供更具体的注释,而不仅仅是基于位点区域和基因。如果你熟悉外显子联盟这个数据库EXAC(ExAC.r0.3.sites.vep.vcf.gz),你可以下载它所有的突变记录数据,看看它对每个变异位点到底注释了些什么,它就是典型的用VEP来注释的。 Continue reading
这个软件比较重要,尤其是对做遗传变异相关研究的,很多人做完了snp-calling后喜欢用ANNOVAR来进行注释,但是那个注释还是相对比较简单,只能得到该突变位点在基因的哪个区域,那个基因这样的信息,如果想了解更具体一点,就需要更加功能化的软件了,snpEFF就是其中的佼佼者,而且是java平台软件,非常容易使用!而且它的手册写的非常详细:http://snpeff.sourceforge.net/SnpEff_manual.html Continue reading
官网:http://mutationassessor.org/
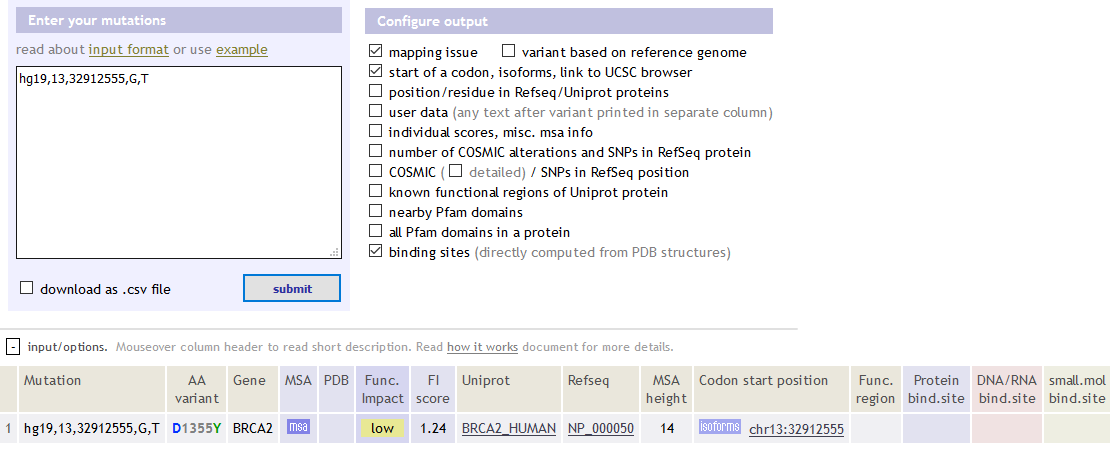
hg19,13,32912555,G,T BRCA2
hg18,7,55178574,G,A GBM
7,55178574,G,A GBM
or in protein space: <protein ID> <variant> <text>, where <protein ID> can be :
1. Uniprot protein accession (i.e. EGFR_HUMAN)
2. NCBI Refseq protein ID (i.e. NP_005219)
examples:
EGFR_HUMAN R521K
EGFR_HUMAN R98Q Polymorphism
EGFR_HUMAN G719D disease
NP_000537 G356A
NP_000537 G360A dbSNP:rs35993958
NP_000537 S46A Abolishes phosphorylation
ID types can be mixed in one list in any way.
感觉最近接触的生物信息学知识越多,越对大数据时代的到来更有同感了。现在的研究者,其实很多都可以自己在家里做了,大量的数据基本都是公开的, 但是一个人闭门造车成就真的有限,与他人交流的思想碰撞还是蛮重要的。
我以前也是用这样的流程 SNP Pipeline Commands 1. Index the reference genome using bwa index /software/bwa-0.7.10/bwa index /reference/japonica/reference.fa 2. Align the paired reads to reference genome using bwa mem. Note: Specify the number of threads or processes to use using the -t parameter. The possible number of threads depends on the machine where the command will run. /software/bwa-0.7.10/bwa mem -M -t 8 /reference/japonica/reference.fa /reads/filename_1.fq.gz /reads/filename_2.fq.gz > /output/filename.sam 3. Sort SAM file and output as BAM file java -Xmx8g -jar /software/picard-tools-1.119/SortSam.jar INPUT=/output/filename.sam OUTPUT=/output/filename.sorted.bam VALIDATION_STRINGENCY=LENIENT CREATE_INDEX=TRUE 4. Fix mate information java -Xmx8g -jar /software/picard-tools-1.119/FixMateInformation.jar INPUT=/output/filename.sorted.bam OUTPUT=/output/filename.fxmt.bam SO=coordinate VALIDATION_STRINGENCY=LENIENT CREATE_INDEX=TRUE 5. Mark duplicate reads java -Xmx8g -jar /software/picard-tools-1.119/MarkDuplicates.jar INPUT=/output/filename.fxmt.bam OUTPUT=/output/filename.mkdup.bam METRICS_FILE=/output/filename.metrics VALIDATION_STRINGENCY=LENIENT CREATE_INDEX=TRUE MAX_FILE_HANDLES_FOR_READ_ENDS_MAP=1000 6. Add or replace read groups java -Xmx8g -jar /software/picard-tools-1.119/AddOrReplaceReadGroups.jar INPUT=/output/filename.mkdup.bam OUTPUT=/output/filename.addrep.bam RGID=readname PL=Illumina SM=readname CN=BGI VALIDATION_STRINGENCY=LENIENT SO=coordinate CREATE_INDEX=TRUE 7. Create index and dictionary for reference genome /software/samtools-1.0/samtools faidx /reference/japonica/reference.fa java -Xmx8g -jar /software/picard-tools-1.119/CreateSequenceDictionary.jar REFERENCE=/reference/japonica/reference.fa OUTPUT=/reference/reference.dict 8. Realign Target java -Xmx8g -jar /software/GenomeAnalysisTK-3.2-2/GenomeAnalysisTK.jar -T RealignerTargetCreator -I /output/filename.addrep.bam -R /reference/japonica/reference.fa -o /output/filename.intervals -fixMisencodedQuals -nt 8 9. Indel Realigner java -Xmx8g -jar /software/GenomeAnalysisTK-3.2-2/GenomeAnalysisTK.jar -T IndelRealigner -fixMisencodedQuals -I /output/filename.addrep.bam -R /reference/japonica/reference.fa -targetIntervals /output/filename.intervals -o /output/filename.realn.bam 10. Merge individual BAM files if there are multiple read pairs per sample /software/samtools-1.0/samtools merge /output/filename.merged.bam /output/*.realn.bam 11. Call SNPs using Unified Genotyper java -Xmx8g -jar /software/GenomeAnalysisTK-3.2-2/GenomeAnalysisTK.jar -T UnifiedGenotyper -R /reference/japonica/reference.fa -I /output/filename.merged.bam -o filename.merged.vcf -glm BOTH -mbq 20 --genotyping_mode DISCOVERY -out_mode EMIT_ALL_SITES