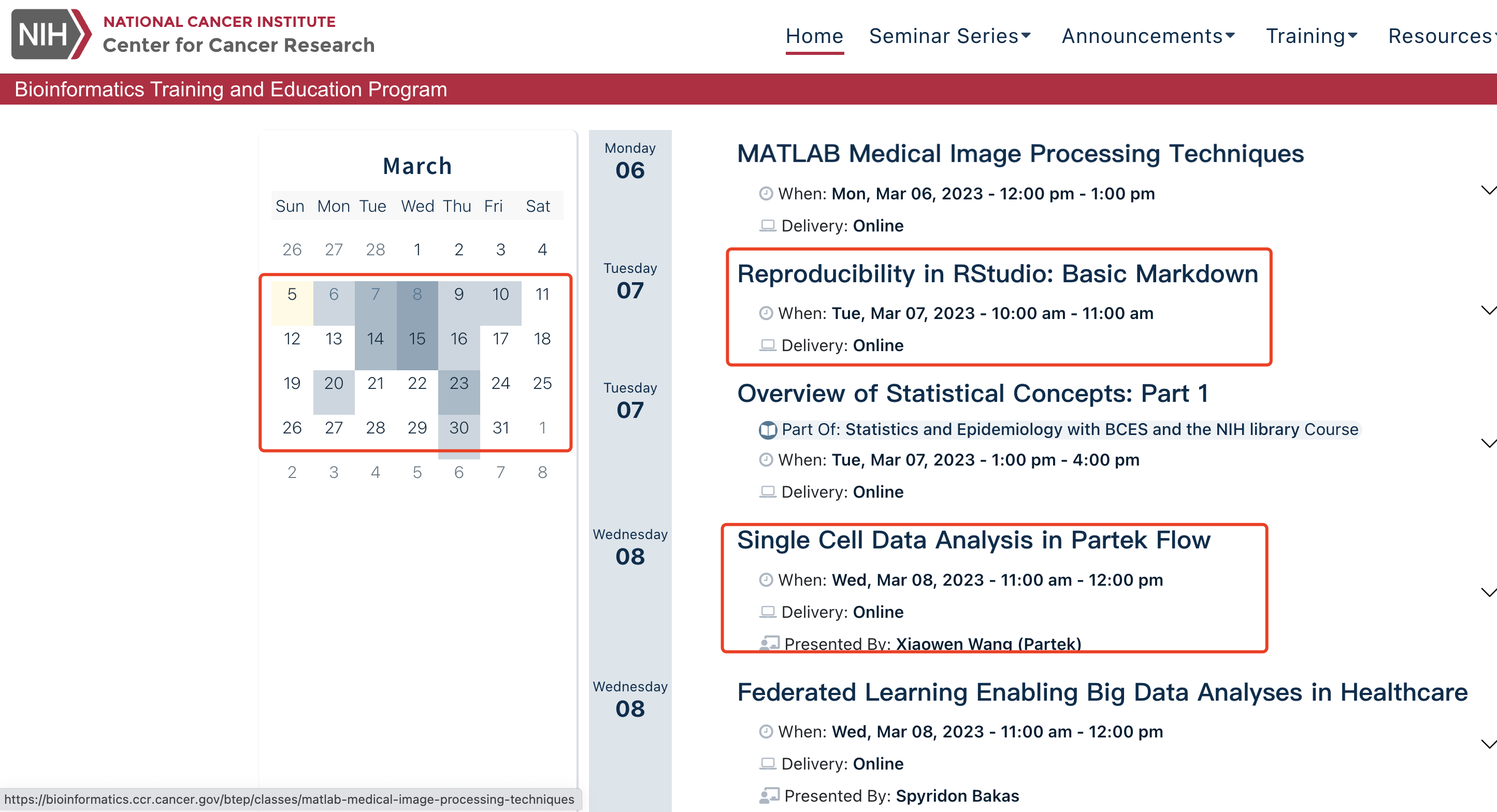
看到了2022的一个单细胞文章:《Single-Cell RNA Sequencing Reveals the Temporal Diversity and Dynamics of Cardiac Immunity after Myocardial Infarction》,里面有表型性状特异性单细胞亚群的概念,详见:Characterization of MI Contribution in Different Macrophage Subsets,需要同时考虑伴随表型变化的单细胞亚群数量以及表达量差异,看起来是蛮复杂的数学公式,如下所示: Continue reading
四
03